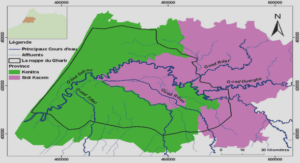
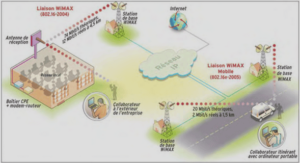
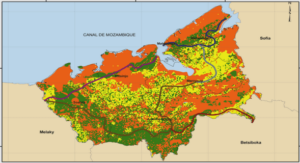
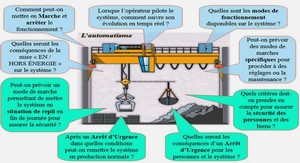
Synthèse et caractérisation par spectroscopies infrarouge, RMN, mössbauer et par diffraction des rayons X
Les composés organostanniques
Généralités
Dans les composés organnostaniques, l’atome d’étain est divalent, trivalent ou tétravalent. Mais étant donné que les orbitales 5d vides peuvent être impliquées dans l’hybridation, un nombre de coordination supérieur à quatre est également possible. La réaction entre un chlorure d’alkylétain et un nucléophile approprié conduit à des alcoolates, carboxylates, amides, thioalkoxides, etc… d’alkylétain. La présence de groupes électronégatifs sur l’étain, le rend susceptible de recevoir des doublets (liaisons datives) des bases de Lewis. Une simple coordination tétraédrique est alors l’exception plutôt que la règle. Dans les Fig. 1.4 et 1.5, sont donnés les principaux environnements possibles autour de l’atome d’étain, aussi bien pour Sn(II) que pour Sn(IV) en supposant que dans le cas de Sn(II), le doublet est également impliqué dans l’hybridation. Fig. 1.4 : Principaux environnements possibles pour Sn(II) Fig. 1.5 Principaux environnements possibles pour Sn(IV). La synthèse du diiodure de diéthylétain par Frankland en 1849 [51] a été le début d’une nouvelle ère dans le domaine de la chimie de l’étain. Toutefois, entre 1849 à 1900, trente-sept articles seulement, relatifs aux composés organostanniques ont été publiés. Krause et Grosse ont réalisés des travaux intéressants sur les composés organostanniques qui furent les premiers à être publiés dans « Organometallische Chemie » [52], en 1937. Ils ont étudié des exemples de tétra alkyl et tétraarylétain ainsi que des halogénures, hydrures, carboxylates, hydroxydes, oxydes, alcoolates et phénolates d’organoétain. En 1960, les travaux sur les composés organostanniques se multiplient avec des illustrations complètes de Ingham et. al., [53]. Yamamoto a publié une brochure comprenant vingt-sept documents sur les composés organostanniques et leurs méthodes de synthèse [54]. Un livre de vingt volumes est publié par Gmelin (certains uniquement sur les composés de l’étain) donne un aperçu global des composés spécifiés (1975-1993) [55-61]. Il existe une source utile de références pour la synthèse, les propriétés, les réactions d’une sélection d’environ mille composés organostanniques dans le dictionnaire des composés organométalliques [62]. Un travail de plusieurs auteurs, édité par Sawyer [63] a été publié en 1991 et les progrès réalisés au cours de la décennie 1970-80 ont été revus par Davies et Smith [64]. En 1989, Saxena et Huber [65] enrichissent la littérature par la mise en évidence de l’activité biologique et de la nature anticancéreuse de la plupart des composés étudiés. La même année, une liste de composés bioorganotétain(IV) fut inventoriée par Molloy et Hartley [66]. Une liste complète de rapports traitant des applications des composés organostanniques dans l’agriculture a été examinée par Crowe [67]. Pellerito et coll [68], ont signalé l’activité biologique des complexes organoétain(IV). De nouvelles procédures expérimentales sont utilisées dans des études biologiques tandis que Barbieri et al., [69] avaient portés essentiellement leurs études sur les interactions des complexes organétain (IV) avec l’ADN et ses dérivés. Blunden et al., [70] et Crow et al., [71] ont publiés des études détaillées sur les applications des organétains(IV) dans la préservation du bois. Bruce [72] a donné une liste exhaustive de composés organostanniques avec leurs structures déterminées par diffraction d’électrons ou des rayons X. Tiekink [73], Holloway et Melink [74] ont axé leurs travaux sur la chimie structurale des composés organostanniques avec plus de quatre cents références. Des techniques de la RMN (119Sn), entre autres, sont utilisées par Martin et al, [75] pour la détermination de la structure des composés de l’étain en solution. Mazhar et al., [76] ont étudié plus de trois centaines de carboxylates organostanniques. Ces travaux ont permis de mettre en évidence les diverses méthodes de préparation des carboxylates organostanniques, leurs propriétés physiques et chimiques et les derniers développements dans la détermination de la structure moléculaire de ces composés. La Royal Society of Chemistry a publié dans des rapports spéciaux, périodiques des listes de toutes les structures qui ont été déterminées par les méthodes de diffraction des rayons x ou d’électrons, jusqu’en 1990 [77]. Le plus important développement ces dernières années a été, cependant, l’utilisation croissante des organostanniques comme réactifs ou comme produits intermédiaires dans la synthèse organique [78,79]. Les composés organométalliques de l’étain (IV), ou composés organostanniques ont au moins une liaison covalente C–Sn. Ces composés contiennent comme centre métallique, un atome d’étain (Sn) tétravalent. Leur formule générale peut s’écrire sous la forme RnSnX4-n, avec n variant de 1 à 4. Selon le nombre d’atomes de carbone liés à l’atome d’étain, les organoétains sont classés en mono-, di-, tri- et tetraorganoétains (RSnX3, R2SnX2, R3SnX, R4Sn). R désigne un groupe alkyle ou aryle et X est une espèce anionique (halogénure, oxyde, hydroxyde, carboxylate ou thiolates) ou un groupe rattaché à l’étain grâce à un atome d’oxygène, de soufre, d’azote, d’halogènes, etc… La synthèse du premier composé organostannique a eu lieu à 1849 par Frankland. Il s’agit du diiodure de diéthylétain, Et2SnI2 [51] suivant l’équation : 2EtI + Sn 150200C Et2SnI2 (1.1) Cependant, avec le début de la commercialisation d’organoétains utilisés comme stabilisant du PVC en 1940, la chimie des composés organostanniques connût un véritable essor et dès lors, des études approfondies sont menées dans ce domaine. La liaison Sn-C est moins rigide que les liaisons C-C ou Si-C, mais elle plus stable en présence d’air et d’humidité ainsi qu’en présence de nombreuses espèces nucléophiles en raison de son caractère non polaire. Alors, il peut y’avoir une rupture de la liaison Sn-C par des réactifs tels que les halogènes, les halogénures métalliques, les acides minéraux, les alcalins, etc… R4Sn + Cl2 R3SnCl + RCl (1.2) R4Sn + SnCl4 2R2SnCl2 (1.3) R4Sn + HCl R3SnCl + RH (1.4) La rupture des groupes aryle, allyle ou vinyle se produisent plus facilement que les groupements alkyles. Aussi, les groupes alkyles à courte chaîne carbonée se décomposent plus facilement que les groupements alkyles dont le nombre d’atomes de carbone est plus élevé. En raison de la grande taille de l’atome d’étain et de la disponibilité des orbitales atomiques de faible énergie 5do vide, un nombre de coordination supérieur à quatre est fréquemment rencontré dans les structures des composés organostanniques. Les applications des composés organostanniques dans l’industrie sont nombreuses et variées [52-54]. Elles dépendent de la stabilité de la liaison de la Sn-C, de la labilité de l’espèce X et de la possibilité d’augmentation du nombre de coordination qui peut être supérieur à quatre. Ces caractéristiques contribuent en grande partie dans leurs applications dans différentes transformations chimiques comme les réactions d’addition sur des composés non saturés ou en tant que catalyseurs et stabilisants du PVC. Les composés organostanniques peuvent être synthétisés par les méthodes standards suivantes. Grignard: SnCl4 + 4RMgCl R4Sn + 4MgCl2 (R = alkyl, aryl) (1.5) Organo Al: 3SnCl4 + 4R3Al 3R4Sn + 4AlCl3 (R = alkyl) (1.6) Direct (Rochow): Sn + 2RX R2SnX2 (R = alkyl) (1.7) Toutes ces voies sont utilisées à l’échelle industrielle tandis que la méthode de Grignard est plus pratiquée au laboratoire. Par contre, il existe une voie de synthèse moins utilisée qui est la réaction de Würtz modifiée. Chapitre 1. Etat de l’art 25 SnCl4 + 4RCl 8NaR4Sn + 8NaCl (1.8) La substitution de R4Sn en un dérivé partiellement halogéné est facilement obtenue en pratiquant la réaction avec le SnCl4. On note une réduction de RnSnX4-n avec LiAlH4 conduisant aux hydrures correspondants. Ceci est une voie intéressante pour les composés organostanniques non symétriques ou hétérocycliques. 1.2.2 Les carboxylates d’organoétain (IV) Ces types de composés organostanniques sont constitués d’entités contenant au moins une liaison Sn-O formée par le biais du groupe carboxylate COO- et peuvent être classés en 3 catégories : Les carboxylates de triorganoétain (IV) ; Les carboxylates de diorganoétain (IV) ; Les carboxylates de monoorganoétain (IV). Les composés carboxylates de di- et triorganostanniques ont de vastes applications dans l’industrie et l’agriculture. Ces composés présentent également un certain nombre de caractéristiques structurales intéressants en raison des divers modes de coordination du ligand carboxylate vis-à-vis de l’étain (IV) [80]. Par conséquent, les deux premières catégories, ont été étudiées plus en détail. Les carboxylates organostanniques sont généralement préparés par des réactions entre des hydroxydes ou oxydes organostanniques et des acides carboxyliques. Ils peuvent s’obtenir également par réaction entre des halogénures organostanniques et des carboxylates métalliques. On note dès fois la rupture d’une liaison carbone-étain en présence d’un acide carboxylique. L’estérification des acides carboxyliques avec des oxydes ou hydroxydes organostanniques est obtenue par déshydratation azéotropique des réactifs dans le toluène ou le benzène, à l’aide du séparateur de DeanStark [81-83]. Pour les composés aryltin(IV), des techniques sont utilisées pour éviter le risque rupture de la liaison SnCAryl)
Les éléments de transition et leurs complexes
Propriétés chimiques des éléments de transition
Tous ces éléments sont des métaux. Ils sont presque tous durs avec des températures de fusion et d’ébullition élevées et un maximum vers le milieu de la ligne. Ils conduisent bien la chaleur et l’électricité. Certains sont des aimants (fer, cobalt, nickel). Ils forment de nombreux alliages entre eux et avec d’autres métaux, notamment avec les lanthanides. La plupart d’entre eux sont attaqués par les acides mais il existe aussi des métaux « nobles » non attaqués (Cu, Ag, Au). Leur chimie est déterminée par la sous-couche d. Les énergies d’ionisation sont relativement faibles et donc les degrés d’oxydation sont nombreux. Ils peuvent monter jusqu’à 8 dans la deuxième et la troisième ligne (Ru, Os). Les électrons d sont très sensibles à l’environnement chimique. Les niveaux d’énergie, dégénérés dans l’ion libre, vont être séparés dans un environnement cristallin (chimie du solide) ou moléculaire (complexes). L’environnement des voisins est appelé sphère de coordination. La levée de dégénérescence est déterminée par la symétrie du site (octaédrique, tétraédrique, …). Son importance augmente de la première ligne à la troisième. De nombreuses géométries sont possibles car la sphère de coordination est souvent très plastique. La description de ces phénomènes peut se faire de manière purement ionique (modèle du champ cristallin) ou moléculaire (théorie du champ des ligands). Le premier modèle est plus simple et rend bien compte des phénomènes de symétrie. La théorie des groupes trouve là une application remarquable (groupes ponctuels de symétrie Oh, Td, D4h ; label de symétrie des orbitales moléculaires dans ce contexte (Oh : t2g et eg ; Td : e et t2; etc.). Le second repose sur la théorie des orbitales moléculaires, qui utilise pleinement la théorie de la symétrie, combinée avec la représentation en termes de moments angulaires (l, s, j pour un électron, L, S, J pour le complexe). Il représente mieux la structure électronique (termes de l’ion libre, obtention des états avec l’introduction du couplage entre le spin et orbite) et de l’extrême versatilité de la liaison de coordination, liaison métal-ligand. Cette liaison peut être décrite simplement d’abord comme un phénomène acido-basique, donneuraccepteur (métal acide de Lewis, ligand donneur). Les interactions peuvent être de symétrie s ou p (en simplifiant), la grande variété des orbitales moléculaires des ligands (électronégativité des donneurs, symétrie des orbitales), la variété des approches métal-ligand, donnent lieu à des modes de liaison très subtils, particulièrement utiles en catalyse, hétérogène ou homogène : ligands s donneurs et/ou p accepteurs ou donneurs. On distingue généralement la chimie de coordination, qui comporte les éléments aux degrés d’oxydation positifs et la chimie organométallique qui comporte les éléments aux degrés d’oxydation nuls ou négatifs. Leur sous-couche d incomplète et le principe de Hund (l’état le plus stable est celui de plus haute multiplicité de spin) permet l’existence de composés magnétiques extrêmement divers, avec un spin maximum S=5/2 pour le manganèse (II) ou le fer(III). En solution aqueuse, les ions des éléments de transition donnent lieu à une chimie très colorée. Les équilibres acidobasiques, d’oxydo-réduction, de précipitation, les potentiels d’électrode sont au cœur de la chimie analytique. Les substitutions de ligands, les échanges d’électrons sont au cœur de la cinétique et de la catalyse.
Les complexes des métaux de transition
Ils peuvent être classés en trois catégories : Les complexes de coordination, très nombreux, degrés d’oxydation positifs, avec des ligands azotés, oxygénés, halogénures … Mononucléaires et polynucléaires De coordination variable (nombre de ligands, et géométrie) Polycations et polyanions (polyoxométallates à degrés d’oxydation élévés) Les complexes organométalliques, très nombreux avec des degrés d’oxydation faibles (0), ligands avec des ligands carbonés (CO, alcènes, alcynes) ou des phosphines permettant des liaisons p de type donneur-accepteur. Mononucléaires et polynucléaires Métaux carbonyles Métallocènes Clusters métalliques Les complexes d’intérêt biologique, métalloprotéines Tous ces composés font l‘objet d’intenses recherches et de nombreuses applications. De nombreux outils théoriques ont été forgés soit pour les décrire (structure, structure électronique, nombre d’électrons) soit pour analyser en détail leurs propriétés chimiques (réactivité, catalyse), physiques et spectrochimiques (spectroscopie électronique, RPE, magnétisme).
Les lanthanides et leurs complexes
Les lanthanides sont les 15 éléments de la première période du bloc f, allant du lanthane (Z = 57) au lutécium (Z = 71). Le lanthane, de configuration électronique [Xe] 5d16s 24f 0 est inclus par extension. Les autres lanthanides adoptent majoritairement la configuration [Xe] 6s24f n+1 . La perte d’un électron 4f et des deux électrons 6s conduit à l’ion lanthanide trivalent qui est le degré d’oxydation le plus stable pour toute la série. C’est à ces ions lanthanides (notés Ln(III) ou Ln3+) que nous nous intéresserons dans ce document.
Propriétés chimiques des lanthanides
Les lanthanides possèdent un même cortège électronique externe (ou valence), qui leur confère une cohérence dans le comportement physique et chimique. Leur configuration électronique correspond à celle du gaz rare xénon, à laquelle des électrons s’ajoutent sur les orbitales 4f, 6s et 5d. Ainsi, le remplissage de l’orbitale interne 4f croît régulièrement du lanthane jusqu’au lutétium A l’état oxydé, les lanthanides n’ont plus d’électrons sur l’orbitale 5d, tout changement dans le nombre d’électrons se reflétant dans l’orbitale 4f. En effet, le changement de configuration électronique se produit plutôt dans les orbitales internes que dans les orbitales externes. D’autres propriétés que nous présentons ci-dessous découlent principalement de la configuration électronique des lanthanides, c’est-à-dire du remplissage de l’orbitale 4f. Les lanthanides sont généralement trivalents dans les conditions naturelles, à l’exception du cérium et de l’europium qui peuvent prendre en plus un autre état ionique. Les proportions des différents états d’oxydation de l’europium ou du cérium sont dépendantes de la composition du milieu, des conditions d’oxydoréduction, de la température et de la pression. Le cérium est tétravalent en milieu oxydant. La réaction Ce3+ → Ce4+ entraîne une diminution du rayon ionique d’environ 15 %. Le rayon ionique des lanthanides est lié au remplissage de l’orbital 4f. Il diminue régulièrement le long de la série, pendant que le nombre d’électrons sur l’orbital 4f augmente. Cette réduction est appelée « contraction lanthanidique ».
Les complexes des lanthanides
Les polymères de coordination à base de terres rares sont généralement préparés à partir de ligands organiques rigides et de faible denticité. Les lanthanides montrant peu de préférence quant à leur nombre ou géométrie de coordination, la structure finale du complexe reste, dans ces cas, très difficile à prévoir, empêchant de ce fait une étude systématique de la relation structure-propriétés. Par ailleurs, la faible denticité du ligand conduit souvent à la coordination de molécules de solvant, pouvant résulter à la désactivation de la luminescence et à une plus faible stabilité thermique liée aux changements structurels considérables associés au retrait de ces molécules. La complexation des éléments f avec des ligands multidentates a été largement étudiée pour l’introduction sélective des cations métalliques au sein d’architectures monométalliques ou polymétalliques discrètes pour le développement de sondes luminescentes solubles dans l’eau. La complexation des ions lanthanides avec des ligands multidentates possédant des chromophores pouvant efficacement absorber l’énergie et la transférer au centre métallique («effet d’antenne») permet d’améliorer grandement l’intensité d’émission des métaux. En effet, la haute denticité du ligand permet de protéger le centre métallique des oscillateurs O-H provenant des molécules de solvant coordinées ou diffusant en première sphère de coordination, et qui conduisent à une désactivation non- radiative des états excités du métal. Des ligands prédisposés, flexibles et multidentates, dans lesquelles un espaceur permet l’orientation d’unités chélatantes, ont déjà été utilisés pour l’élaboration contrôlée de complexes monométalliques.
Avant-propos |