Séquençage du génome complet – WGS (Whole Genome Sequencing)
L’obtention de la totalité de l’information génomique d’un isolat fournit théoriquement une méthode de typage offrant la meilleure résolution (Ruppitsch, 2016). Avec le développement des technologies de séquençage haut débit dans les années 2000, il devient de plus en plus abordable et rapide de recourir au séquençage du génome complet pour le typage des souches de Salmonella (Ruppitsch, 2016). L’emploi du WGS comme méthode de routine pour l’analyse des souches isolées permet une augmentation constante du nombre de génomes de Salmonella disponibles dans les bases des données en libre accès. Disposer d’un grand nombre de génomes permet une meilleure utilisation des données issues du séquençage pour la source-attribution (Dunn, 2016). Les méthodes d’analyse génomique basées sur le WGS ont été rendues possible par le développement des techniques de séquençage à haut débit dites « short reads » de 3éme génération telles que celles proposées par la technologie Illumina. La figure 10 détaille le fonctionnement de cette technologie. L’ADN génomique est découpé en fragments courts sur lesquels des adaptateurs sont fixés afin d’être amplifiés par PCR et créer une librairie de séquences. Les séquences de chaque librairie sont ensuite fixées via d’autres adaptateurs sur une phase solide puis séquencées à l’aide de bases marquées émettant des signaux lors de leur Une fois les séquences génomiques obtenues, de nombreux types d’analyses sont envisageables. L’étude de la diversité génétique d’une collection de souches repose le plus souvent sur des techniques de variant calling qui consistent à détecter le polymorphisme analysées (Timme et al., 2013). La recherche des variants (variant calling) permet d’étudier les relations entre des souches éloignées et proches génétiquement et a été utilisée avec succès dans l’investigation des TIAC (Timme et al., 2013; Allard et al., 2016; Dunn, 2016; Ruppitsch, 2016). Le séquençage de l’ADN génomique donne également accès à des informations telles que le profil MLST d’une souche, son résistome (ensemble des gènes de résistance d’une souche donnée), mobilome (l’ensemble des éléments mobiles tel que les plasmides ou les transposons d’un génome) ou encore le profil de facteurs de virulence (Taboada et al., 2017).
Différents outils permettent la prédiction d’informations d’intérêt tel que le profil MLST d’une souche, le sérotype d’une souche à partir de son génome (application SISTR/SeqSero) (Yoshida et al., 2016), la prédiction de la présence de gènes de résistance à certaines classes d’antibiotiques (Zankari et al., 2012) mais également une annotation fonctionnelle rapide et la prédiction de caractéristiques phénotypiques (Page et al., 2015) et des études d’association basées sur le génome complet (Genome Wide Association Study ou GWAS) (Brynildsrud et al., En Europe, 18 pays utilisent le WGS pour l’investigation de TIAC et l’ECDC encourage la transition vers une surveillance basée sur le WGS dans l’espoir de garantir une meilleure couverture des souches collectées qui dépassent actuellement la capacité européenne de Toutefois le séquençage du génome complet possède quelques inconvénients : cette approche impose des dépenses nouvelles pour assurer le séquençage et demande une expertise en bio-informatique dans les laboratoires de référence. De même, une puissance de calcul importante doit être dédiée à l’analyse de jeux de données et au stockage des données issues du séquençage. Il est également nécessaire de standardiser les méthodes de traitement des données et d’analyse de la diversité génétique afin de pouvoir la comparer aux méthodes traditionnelles (Ellington et al., 2017; Ferrari et al., 2017). Le groupe de travail 25 (ISO/TC 34/SC 9/WG 25) de l’Organisation Internationale de Standardisation (ISO) travaille actuellement sur une norme sur la génération de données WGS et sur les techniques d’analyses à appliquer sur ces données.
Les facteurs moléculaires permettant l’infection entérique et systémique par Salmonella
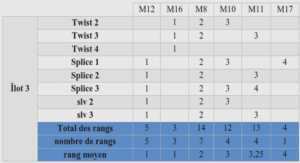
Après l’ingestion, la virulence d’une souche de Salmonella varie beaucoup d’un sérovar à l’autre. Toutes les salmonelles doivent cependant surmonter les mêmes obstacles : elles doivent traverser l’estomac et trouver le moyen de persister dans le milieu hautement compétitif de l’intestin pour traverser l’épithélium intestinal et infecter l’hôte. La virulence d’une souche de Salmonella dépend de nombreux facteurs. Elle est déterminée par sa capacité à adhérer aux cellules, à les envahir, à survivre et à se multiplier à l’intérieur des cellules épithéliales et des macrophages. Pour le succès de ces trois étapes clés (adhésion, invasion, multiplication intracellulaire) de nombreux gènes vont intervenir. Une grande partie de ces gènes de virulence est regroupée dans des régions génomiques spécifiques, acquises par transfert horizontal, appelées « Ilots de pathogénicité de Salmonella » (SPI) (Marcus et al., 2000; de Jong et al., 2012; Hurley et al., 2014). L’ensemble de ces gènes de virulence va permettre la colonisation de l’hôte et l’invasion systémique de Salmonella.