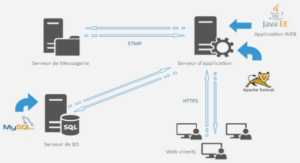
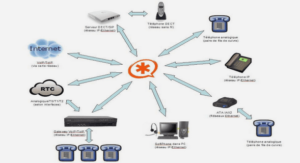
Télécharger le fichier original (Mémoire de fin d’études)
Théorème
Le second théorème fondamental de la DFT dit que la fonctionnelle de l’énergie totale d’un système à N électrons possède un minimum correspondant à l’énergie de l’état fon-damental et à la densité électronique de l’état fondamental [1]
= ⁄ >
E0 [fl(r)] = min F [fl(r)] + Vext(r)fl(r)d3r .
Ce théorème utilise le principe variationnel i.e. on minimise l’énergie sous la contrainte d’orthogonalité de la base ÈÂi|ÂjÍ = ”ij donnant le système d’équations dites de Kohn-Sham
S ~2 fl(rj) T
W≠ Ò2 + ⁄ d3rj + Vxc(r) + Vext(r) ≠‘i X Âi(r) = 0, (II.4)
2m ri ≠ rj |
W | X
W X
W X
U ¸ ˚˙ ˝ V
Veff(r)
pour chaque orbitale Âi appelés orbitales de Kohn-Sham. La résolution de cette équation requiert la connaissance du potentiel effectif Veff(r). On peut alors obtenir la densité fl(r) = 2 qi fi|Âi(r)|2 où le facteur 2 tient compte de la dégénérescence de spin et le terme fi est l’occupation de l’orbitale i (0 Æ fi Æ 1). Le potentiel d’échange-corrélation est alors obtenu par différenciation de l’énergie d’échange-corrélation par la densité
Vxc(r) = ”Exc[fl(r)].
”fl(r)
Ce potentiel obtenu est local, il ne dépend pas de la valeur du potentiel en d’autres points de l’espace et est alors égal à Vxc[fl(r)]. Un nouveau potentiel effectif peut être calculé, il dépend de la densité électronique qui elle-même dépend des fonctions d’ondes mono-électroniques Âi, le processus est dit auto-cohérent [5].
On peut remarquer que la localité du terme d’échange-corrélation rend les équations de Kohn-Sham moins complexes que les équations de Hartree-Fock, pouvant être obtenues par un principe variationnel similaire. Ces dernières comprennent seulement le terme
THÉORIE DE LA FONCTIONNELLE DE LA DENSITÉ
d’échange non-local qui est difficilement évaluable. Si on connaît la forme exacte de Exc, alors l’énergie totale du système est bien la valeur propre de l’opérateur hamiltonien de l’équation de Schrödinger et est donc considérée comme exacte. L’approximation se fait sur le choix de l’énergie d’échange-corrélation et du potentiel d’échange-corrélation. Il existe plusieures fonctionnelles dont le but est de décrire au mieux l’interaction d’échange-corrélation inconnue, le choix dépend du système à étudier.
Les orbitales de Kohn-Sham n’ont pas de signification physique, en revanche la somme de leur carré décrit la densité exacte. Le déterminant de Slater construit sur les orbitales de Kohn-Sham n’a donc rien à voir avec la fonction d’onde multi-électronique. Les ap-proximations faites dans les calculs DFT permettent de simuler des systèmes de plus grandes tailles comparé à la méthode de Hartree-Fock.
Fonctionnelles d’échange-corrélation
Ces fonctionnelles comprennent toutes les contributions inconnues du problème à N corps. Elles incluent les effets d’échange et de corrélation exposés plus haut mais aussi les effets non-classiques dûs à la self-interaction et à l’énergie cinétique. Il existe différents degrés d’approximation du potentiel d’échange-corrélation allant du moins précis au plus précis aboutissant à la fonctionnelle exacte.
L’approximation de la densité locale (LDA) est basée sur un gaz d’électrons uniformé-ment réparti dans l’espace. Ce dernier est subdivisé en éléments de volume infinitésimaux dans lesquels la densité est constante
ExcLDA[fl(r)] = ⁄ fl(r)‘xc fl(r) » dr.
!
La densité d’énergie ‘xc(fl) est alors exprimée comme la somme des contributions d’échange et de corrélation. Pour un gaz d’électrons uniforme, l’énergie d’échange est connue de ma-nière exacte. L’énergie de corrélation peut être obtenue par d’autres méthodes comme des méthodes Monte-Carlo. Une expression a été proposée par Vosko, Wilk et Nusair en 1980 [6]. Cette approximation, bien que ne comportant aucun paramètre empirique, manque globalement de précision. Lorsqu’on utilise ce type de fonctionnelle sans ajout de termes correctifs, il s’agit de calculs premier principe.
L’approximation du gradient généralisé (GGA) consiste à ajouter à la LDA une quan-tité non-locale qui est le gradient de la densité. Elle prévoit une distribution électronique non-homogène où la plus forte densité électronique est située proche des noyaux ato-miques. Cette addition peut être vue comme le développement en série de Taylor au premier ordre de l’énergie d’échange-corrélation par rapport à la densité électronique [7]
⁄
ExcGGA[fl(r)] = fl(r)‘xc!fl(r), Òfl(r) » dr.
Ces fonctionnelles sont formées à partir de paramètres ajustés sur des données expéri-mentales avec une grande précision et ne font plus vraiment partie des méthodes premier principe par l’apport de ces données empiriques.
Parmi les fonctionnelles GGA, nous avons employé dans cette thèse la fonctionnelle de Perdew, Burke et Ernzerhof (PBE) [8]. Cette fonctionnelle n’utilise pas de données expérimentales et est connue pour sa bonne précision sur un large domaine de systèmes.
Fonctionnelles hybrides
L’utilisation des orbitales de Kohn-Sham pose problème dans l’application de la DFT en matière condensée. L’un des problèmes les plus connus dans les calculs premier principe est la sous-estimation du gap dans les semi-conducteurs et les isolants, l’erreur pouvant atteindre 50%. Le problème vient de l’absence d’un terme dans les gaps de Kohn-Sham (Egxc) provenant de la discontinuité des dérivés des fonctionnelles d’échange-corrélation [9]. Ce terme est absent par construction de chaque fonctionnelle approximée (LDA, GGA ou autre).
Becke, en 1993, a montré qu’une fraction de l’énergie d’échange exact de Hartree-Fock introduite dans la fonctionnelle premier principe permet une amélioration significative de la précision en DFT. Les fonctionnelles obtenues sont appelées fonctionnelles hybrides. Elles sont construites comme une combinaison linéaire de l’échange exact de Hartree-Fock et de différentes fonctionnelles d’échange-corrélation. Le poids de chaque contribution est ajusté sur des données expérimentales. La fonctionnelle hybride qui connaît le plus de succès est la fonctionnelle B3LYP [10, 11]
ExcB3LYP = ExcLDA + 0.2(ExKS ≠ ExLDA) + 0.72ExB88 + 0.81EcLYP + 0.19EcVWM.
Ex dénote l’énergie d’échange et Ec l’énergie de corrélation. L’énergie d’échange vient du déterminant de Slater de Kohn-Sham et non celui de Hartree-Fock. Dans le cadre GGA, l’échange est apporté par la fonctionnelle de Becke B88 [12] et la corrélation par celle de Lee-Yang-Parr (LYP) [13]. Une partie du potentiel de Vosko-Wilk-Nusair(VWN) [14] est aussi introduite. Cette fonctionnelle hybride est l’une des plus précises et plus populaire mais très coûteuse en ressources de calcul et inapplicable pour des surfaces.
La fonctionnelle hybride de Heyd, Scuseria et Ernzerhof (HSE) évalue l’échange de Hartree-Fock par un potentiel de Coulomb écranté [15]. Elle a l’avantage d’être une des méthodes hybrides les moins coûteuses car l’évaluation de l’échange de Fock est réduite à de petites distances entre électrons. Elle est définie par une fraction de l’échange de Fock a à séparation entre électrons nulle et d’une échelle de longueur Ê≠1 sur laquelle l’échange de Fock courte-portée est calculé
ExcHSE = aExKS,CP(Ê) + (1 ≠ a)ExPBE,CP(Ê) + ExPBE,LP(Ê) + EcPBE,
où LP et CP signifient longue portée et courte portée respectivement. L’échange de Hartree-Fock est calculé en utilisant la matrice densité de Kohn-Sham fl‡,‡Õ (r, rÕ)
ExKS,CP(Ê) = ≠2 ‡,‡ ⁄ r | r r≠Õ | ) |fl‡,‡Õ (r, rÕ)|2 drdrÕ,
1 ÿÕ erfc(Ê rÕ
| ≠ |
avec erfc la fonction erreur complémentaire et r et ‡ sont respectivement les vecteurs positions et les états de spin des électrons.
Formalisme des ondes planes
Pour modéliser nos systèmes, nous avons utilisé le code de dynamique moléculaire VASP-5 (Vienna Ab-initio Simulation Package) basé sur la DFT employant des en-sembles de bases d’ondes planes combinés avec des pseudo-potentiels issus d’ondes planes augmentées (PAW).
Les pseudo-potentiels
Le formalisme des pseudo-potentiels utilise le fait que les propriétés chimiques des molé-cules et des solides sont principalement gouvernées par les électrons de valence. Les élec-trons situés proches des noyaux, aussi appelés électrons de cœur, sont considérés comme chimiquement inertes et leur potentiel associés peut être substitué par un potentiel effectif appelé pseudo-potentiel. Ce dernier n’interagit qu’avec les électrons des couches externes qui, quant à eux, peuvent être traités un par un. Le choix d’un pseudo-potentiel doit être basé sur le caractère des liaisons entre atomes du système. Si le potentiel extérieur est remplacé par un pseudo-potentiel, le hamiltonien décrivant les électrons de valence devient un pseudo-hamiltonien et les fonctions d’ondes sont des pseudo-fonctions d’ondes. L’approximation du pseudo-potentiel consiste à considérer les pseudo-fonctions d’ondes comme des fonctions d’ondes. Les pseudo-potentiels à norme conservée sont tels que la norme de la pseudo-fonction d’onde est identique à la norme de la fonction d’onde. Les pseudo-fonctions d’ondes issues de potentiels dits ultra-doux
Table des matières
I Aspects théoriques
I.1 Propriétés des nitrures
I.1.1 Structure cristalline
I.1.2 Ionicité des liaisons
I.2 Polarité dans les matériaux
I.2.1 Instabilité des matériaux polaires
I.3 Modèles de structure électronique dans les films minces polaires
I.3.1 Matériaux non-polaires
I.3.2 Films polaires
I.3.3 Travail de sortie dans un matériau polaire
I.4 Métaux sur isolant
I.4.1 Croissance
I.4.2 Adhésion
II Méthodes numériques
II.1 Théorie de la fonctionnelle de la densité
II.1.1 Généralités
II.1.2 Fonctionnelles d’échange-corrélation
II.1.3 Formalisme des ondes planes
II.2 Calcul des charges
II.2.1 Méthodes des fonctions de base
II.2.2 Méthodes de la densité électronique
II.2.3 Corrections dipolaires
IIIReconstruction (2 ◊ 2) de l’AlN(0001)
III.1 AlN
III.1.1 AlN(0001)
III.2 Calculs préliminaires
III.2.1 AlN en volume
III.2.2 Surfaces È0001Í d’AlN
III.3 Reconstructions de surfaces polaires
III.3.1 Règle du comptage des électrons
III.3.2 Optimisation des reconstructions de l’AlN(0001)
III.3.3 Critère de stabilité électrostatique
III.3.4 Structure électronique des reconstructions
III.3.5 Stabilité des reconstructions
IV Dépôt de métaux sur la reconstruction (2 ◊ 2)-Nad
IV.1 Dépôt de magnésium
IV.1.1 Observations expérimentales
IV.1.2 Structures atomiques
IV.2 Dépôt d’argent
IV.3 Dépôt d’or
IV.3.1 Observations expérimentales
IV.3.2 Structure atomique de Au/AlN(0001)
IV.3.3 Adhésion de Au/AlN(0001)
IV.3.4 Structure électronique
IV.4 Liens avec l’expérience
IV.4.1 Travail de sortie
IV.5 Conclusion
Conclusion
A Calcul de la constante de Madelung
B Étude d’un complexe de Ru(dbm)2 sur Au(111)
B.1 Introduction
B.2 Étude préliminaire : Ru(dbm)3
B.2.1 PDOS
B.3 Ru(dbm)2 sur Au(111)
B.3.1 Interprétation des images
B.3.2 Calculs sur surface
B.3.3 Effet des atomes de bromes
B.3.4 Conclusion