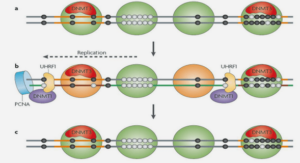
Le système Mismatch Repair ou MMR
Post réplicative, l’activation du MMR est liée à la présence de bases mal appariées (erreur d’incorporation par les ADN polymérases et lors de la réplication) ou hétéroduplexes. La reconnaissance est faite par un hétérodimère associant soit MSH2 à MSH6 soit le couple MSH2 MSH3 en cas de petites insersions-délétions. Puis ces dimères recrutent MSH1 et PMS2. Ce complexe MSH2-MSH6/3-MLH1-PMS2 va glisser sur l’ADN et s’éloigner du mésappariement. Ce groupe va distinguer le brin contenant la base erronée de l’autre brin, précisément où il existe une interruption des brins (fragments d’Okazaki ou du côté 3’OH libre du brin direct). Une exonucléation par EXOI-PCNA suit cette reconnaissance pour dégrader le brin mésapparié. Le brin est alors resynthétisé et une ligase IV rétablit la continuité de l’ADN.
Recombinaisons de l’ADN
La recombinaison permet la réparation des cassures double brin.
Deux molécules senseurs de la famille des PI3K (Phosphatidyl Inositol Kinase related protein Kinase) entrent en jeu : ATM et ATR [Kurz EU, 2004, Shiloh Y, 2003].
On distingue deux grandes voies de recombinaison :
– la recombinaison homologue (RH)
– la recombinaison non homologue des extrémités ou end joining (NHEJ).
La RH se sert, comme modèle, du chromosome homologue contenant la séquence homologue non endommagée [Friedberg EC, 2003, Reliene R, 2007]. ATM vient phosphoryler le complexe MRN (Mre11-Nb1-Rad50) au niveau des extrémités de la cassure [D’Amours D, 2002]. Le complexe est doté d’une activité 5’-3’-exonucléase qui va générer deux extrémités simple brin 3’, immédiatement recouverte par RPA [Hoeijmakers JH, 2001]. La recombinaison commence : RPA est déplacé par Rad51, Rad52 et Rad54, accompagnés de la création de nucléofilaments autour de ces extrémités 3’, en présence de Brca1 et de Brca2. Rad51 possède une activité recombinase qui va permettre la reconnaissance de la séquence homologue sur la chromatide soeur.
La NHEJ permet quant à elle de joindre les extrémités double brin induites par la cassure après lareconnaissance de la lésion [Hoeijmakers JH, 2001, Lieber MR, 2010, Stracker TH, 2004]. Cette jonction est possible par l’intervention d’un gros complexe comprenant la DNA-PK (possédant une sous unité catalytique), des molécules Ku et Artémis. Cette dernière molécule est phosphorylée par la DNA-PK, ce qui va activer sa portion 3’-5-exonucléase afin de bien proposer ces extrémités avant la ligation par le complexe XRCC4-ligase IV pour la restauration de la continuité de la double hélice [Ma Y, 2002].
La réversion directe
Elle permet la réparation, après la création d’une O6 méthylation d’une guanine, par l’action d’une méthylguanine methyltransférase (MGMT). Ainsi, le groupement méthyl de l’O6 guanine est transféré de manière irréversible sur un résidu cystéine de la MGMT. Cet ensemble subit ensuite une protéolyse faisant disparaître l’anomalie [Xu-Welliver M, 2002].
MECANISMES FONDAMENTAUX DE L’ONCOGENESE
Généralités
On distingue classiquement six mécanismes fondamentaux à l’oeuvre dans l’oncogenèse.
On peut y adjoindre l’instabilité génétique, qui permet à la cellule cancéreuse d’explorer avec succès, selon le modèle de la sélection darwinienne, les voies les plus diverses qui lui permettront de subsister, de proliférer et d’envahir les tissus voisins et distants.
Ces six mécanismes fondamentaux sont les suivants :
– indépendance vis-à-vis des signaux de prolifération,
– perte du contrôle du cycle cellulaire,
– perte des capacités d’apoptose,
– acquisition du phénotype d’immortalité des lignées cellulaires,
– développement des capacités d’invasion et de métastase,
– mise en place d’une angiogenèse spécifique à la tumeur.
Les deux derniers mécanismes ne seront pas traités, car ne s’appliquent que peu aux leucémies myéloïdes, étant surtout des tumeurs « liquides ».
L’instabilité génétique passe par l’altération des capacités de réparation de l’ADN. Il existe des altérations génétiques du type « gain de fonction » (oncogènes) ou du type « perte de fonction » (gènes suppresseurs de tumeurs).
Gènes suppresseurs de tumeur et prédispositions aux cancers
Le concept de gène suppresseur de tumeur a été développé à partir du rétinoblastome bilatéral, parfois familial et de l’observation de la délétion du 13q constitutionnelle dans certains de ces cas, contrastant avec les rétinoblastomes unilatéraux et sporadiques. Le mécanisme « à deux coups » décrit par Knudson rend compte de cette observation. Un des deux allèles est altéré dans la totalité des cellules de l’organisme à partir de la lignée germinale. Le second événement survient de façon sporadique sur le deuxième allèle d’une cellule somatique de chaque rétine entraînant la transformation. Cette hypothèse a été validée par les progrès importants de la génétique humaine. Elle explique les syndromes de prédisposition héréditaire aux cancers transmis sur le mode dominant. A chaque gène suppresseur de tumeur est associé un syndrome de prédisposition héréditaire aux cancers, se manifestant parfois simplement par un risque élevé de développer un cancer mais parfois accompagné d’une pathologie caractéristique. On peut citer certains gènes tels BRCA1/BRCA2 et une prédisposition aux cancers héréditaires du sein, NF1 et la neurofibromatose de type 1, TP53 et le syndrome de Li-Fraumeni, WT1 et le néphroblastome parfois associé à une délétion de 11p13 dans les cas familiaux et/ou bilatéraux.
D’autres maladies constitutionnelles de transmission récessives peuvent prédisposer à la transformation maligne telles l’ataxie télangiectasie avec son gène ATM qui présente une instabilité chromosomique au niveau des récepteurs T. L’anémie de Fanconi (AF), avec sa prédisposition aux SMD et aux LAM, est provoquée par des doubles mutations sur au moins 15 gènes possibles: FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ/BRIP1, FANCL, FANCN/PALB2, FANCM, FANCO/RAD51C, FANCP.
Outre un syndrome malformatif et l’anémie grave d’origine centrale, s’y associent des cassures chromosomiques dont certaines figures d’échanges comme des figures quadriradiales ou triradiales. Ces anomalies chromosomiques non systématisées, sont présentes spontanément en culture de sang +PHA de 72h. Elles peuvent être augmentées par une hypersensibilité des cellules d’AF aux agents alkylants bifonctionnels. De plus il existe des anomalies dans les mécanismes de réparation de l’ADN et un passage plus lent des cellules à passer de la phase G2 à la phase M.
Principes des mécanismes d’action d’agents anticancéreux potentiellement leucémogènes
Les différents sous chapitres suivants ne concerneront essentiellement que les agents alkylants, les anti-topoisomérases II et la radiothérapie.
Les Agents Alkylants (AA)
L’alkylation est le transfert d’un groupe alkyl d’une molécule à une autre en créant en général une liaison covalente. Cette réaction permet de rajouter des radicaux chimiques à des molécules cibles. Les agents alkylants utilisés en thérapeutique créent des liaisons covalentes via des groupes alkyl sur différents constituants de la cellule, dont l’ADN qui est considéré comme la cible principale.
La plupart des AA sont des agents méthylants monofonctionnels tels que temozolomide, N-methyl-N’-nitro-N-nitrosoguanidine (MNNG), dacarbazine (voir tableau 2)
Les autres AA sont bifonctionnels tels que les moutardes (chlorambucil, cyclophosphamide, melphalan) ou les agents chloro-ethylants (mustine, carmustine, lomustine et fotemustine) (voir tableau 2).
Les AA monofonctionnels
Mécanismes d’action
Les agents methylants monofonctionnels fixent des radicaux méthyls sur les atomes N et O des bases nucléiques. Cela crée des dommages à l’ADN, de type faux sens, cassure simple brin ou dépurination qui provoquent mutations, cancérogenèse mais surtout mort cellulaire. Les liaisons à partir des N-méthylations représentent plus de 80 % des bases méthylées [Kondo N, 2010]. Les méthylations de ces bases « alkylées » ont différents niveaux de stabilité.
La méthylation en N7-methylguanine (N7MeG) a été identifiée comme la N-méthylation la plus stable et prolongée dans le temps [Beranek DT, 1990]. La O6-méthylation a également un caractère stable [Goth-Goldstein R, 1980 – Kaina B, 1990]. Bien que les O- alkylations soient minoritaires dans les mécanismes d’alkylation, celles-ci sont bien plus génotoxiques et mutagéniques que les N-alkylations. Ces dernières sont quant à elles plus cytotoxiques [Goth R, MF, 1974, Kaina B, 1990]
Mécanismes de réparation de l’ADN utilisés par la cellule
Il existe différents intervenants.
Le système BER va intervenir au niveau de N7MeG, N3MeA et N3MeG. Une DNA glycosylase spécifique, l’alkyladénine DNA glycosylase (Aag) va agir en premier rendant les bases apuriques ou apyrimidiques. Puis, APE1 qui reconnaît ces bases apuriques ou apyrimidiques, va retirer les groupes 3’-OH et 5’-desoxyribosephosphate (5’dRP) cytotoxiques. L’ADN polymérase reconstruit la partie nucléotidique manquante et relie également le groupe 5’dRP. Enfin, la DNA ligase I ou II et XRCC1 termine le processus. Il a été montré qu’en l’absence de la polymérase , le 5’dRP persiste dans la cellule la rendant très sensible au méthylméthanesulfonate [Sobol RW. 1996, Sobol RW. 2002]
Il existe aussi un système homologue AlkB chez l’homme hABH, qui intervient au niveau de N1MeA, N3MeC, N3MeT et N1MeG. [Karina B, 2007]. Cette enzyme a une action oxydative démethylase présente sous deux isoformes hABH2 qui intervient essentiellement au niveau de la fourche de réplication en phase S et la majorité du système repose sur l’isoforme hABH3.
Mécanismes de réparation de l’ADN utilisés par la cellule
Ici aussi, c’est la Méthylguanine DNA méthyltransférase qui va réparer les lésions N1G:N3C entraînées par les nitrosourées.
En revanche, la réparation des lésions interbrins est plus complexe en associant l’action du FA, de NER, de la synthèse trans-lésionnelle et de la RH [Thompson LH, 2009}.
Les protéines FANCM et FANCA associating polypeptide 24 vont former un hétérodimère capable de fixer l’ADN au niveau de la fourche de réplication lorsque celle-ci a été bloquée.
Les protéines XPF et ERCC1 du système NER vont alors couper l’ADN de part et d’autre de la lésion interbrin. Le segment manquant va alors être remplacé par les polymérases du système de synthèse trans-lésionnelle.
Enfin la RH est activée lorsque FANCD2 est ubiquitinée par le complexe FA.
Les anti topoisomérases II (ATII)
Mécanismes d’action
Les ATII empêchent la topoisomérase II de fonctionner lors de son action de religation des 2 brins de l’ADN qu’elle vient de couper. Il en résulte une cassure double brin. On distingue 2 groupes d’ATII : les épipodophyllotoxines représentées par leur chef de file l’étoposide et les anthracyclines telles que l’épirubicine (voir tableau 2) Ces 2 types de molécules induisent plutôt des échanges de matériel nucléosidique équilibrés mais distincts car les points de cassure ne sont pas identiques.
Les épipodophyllotoxines induisent préférentiellement des réarrangements au niveau de la région 11q23 et quelques fois en 21q22 alors que l’épirubicine donne naissance à des translocations t(15;17) propres aux LAM 3.
Le point de cassure 11q23 post étoposide
La plupart des réarrangements du gène MLL (qui mesure 120 Kb) provoque la fusion de celui-ci en 5’avec d’autres gènes partenaires. La zone de fusion représente quant à elle 8,3 Kb.
Ces types de réarrangements sont aussi bien retrouvés dans les LAL que dans les LAM. Les translocations t(9;11), t(6;11) et t(11;19) sont les plus fréquemment affichées par les LAM. L’équipe de Langer [Langer T, 2003] a travaillé sur des leucémies aigues avec une t(9;11) aboutissant à la fusion chimérique AF9-MLL. On comptait parmi 11 patients étudiés, 5 t-LAM post ATII sans avoir reçu une épipodophyllotoxine. Le point de cassure passant par MLL était identique au niveau de la partie télomérique de l’intron 8, au milieu de la région de coupureattachement et très proche du site de fixation de la topoisomérase II (appelé scaffold attachment region ou SAR).
MLL représente un point de cassure visé par l’ensemble des ATII qui n’est pas exclusif au épipodophyllotoxines.
La translocation t(15;17) post épirubicine
L’équipe de Mays [Mays AN, 2010], montre que les points de cassures retrouvés sont localisés aux introns 6, 3 et dans l’exon 7 du gène PML. L’atteinte de l’intron 6 n’est pas la même que celle induite par la mitoxanthrone. Le point de cassure se situe au niveau de l’intron 2 du gène RARA.
L’ensemble de ces points de cassure de la translocation (15;17) est ainsi préférentiellement ciblé par l’épirubicine et ces lésions induites sont stables. Les extrémités en 5’, générées par la topoisomérase II en PML et RARA occasionnent alors la mise en route du système NHEJ des cassures double brin. Les auteurs suggèrent que les cassures double brin dans des régions de susceptibilité aux ATII sont réparées de façon aberrante.
Il est supposé qu’après une aplasie post chimiothérapie, les cellules progénitrices hématopoïétiques vont, chez certains patients, prendre un avantage sélectif lorsqu’elles possèdent cette translocation de type t(15;17).
La translocation t(9;22)
La translocation équilibrée t(9;22) que l’on peut retrouver dans des t-LAM, ne semble pas être spécifique d’un agent inducteur tel que les ATII. Cette translocation peut également être induite par les AA, la radiothérapie ou même le benzène. Ceci laisse suggérer que ces points de cassure en 9q34 et 22q11 ne seraient pas vraiment spécifiques dans un contexte de t-LAM. [Lichtman MA, 2008]