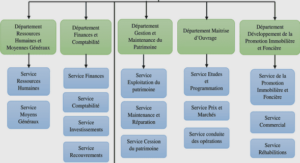
L’approche du couplage fermé (Close Coupling CC)
La description des équations ayant un degré de liberté interne fait toujours appel à l’approche du couplage fermé (Close Coupling CC). Cette approche a été appliquée pour la première fois à la théorie des collisions en 1960 par Arthur et Dalgarno (Arthurs, A. M., & Dalgarno, A. (1960)) entre une molécule BC qui sera considérée comme un rotateur rigide sans vibration en collision inélastique avec un atome A sans structure c’est à dire pas de niveau d’énergie, ce qui fait que son énergie interne est constante.
La fonction d’onde
Lors de la collision, le potentiel d’interaction reste constant et le moment angulaire total J est aussi conservé avant et après la collision. M est la projection de J suivant l’axe z’ telle que J ≤ M ≤ J et dans ce cas: S ( jl , j ‘ l ‘ ) est la matrice de diffusion à partir de laquelle on peut déterminer les sections efficaces et qui est diagonale à J et ne dépend pas de M. Elle renseigne sur le comportement de la molécule en collision avec l’atome par l’intermédiaire du potentiel et est liée à la matrice de transition T ( jl , j ‘ l ‘ ) par la relation suivante: À partir de la matrice de transition T ( jl , j ‘ l ‘ ) on peut déterminer la section efficace totale des sous niveaux de transition j, m→j’, m’ en considérant les éléments de matrice de transition comme une somme d’éléments de matrice réduite (Alexander & Davis (1983)).
Les méthodes d’approximations
L’approximation des états couplés (Coupled StatesCS) Cette approximation a été introduite en 1974 par McGuire et Kouri (Paul McGuire and Donald J. Kouri (1974)) pour l’étude dynamique des molécules. Elle a comme avantage d’augmenter la vitesse de calcul de 10 à 20 % fois plus que celle de CC pour les grandes énergies et avec une très bonne précision. Dans cette étude, au lieu du repère fixe SF on choisit plutôt le référentiel mobile BF(x’,y’ ,z’) où z’ est confondu avec la distance R de telle sorte que la projection de l sur l’axe z’ est nulle et celle de j sur l’axe z’ est notée α. Ainsi on note la fonction du rotateur rigide Yjα j(θ,φ) qui est aussi notée │jα JM> et la fonction d’onde du système dans le repère mobile développée par Launay dans les années 1976 et 1977 s’écrit..
Cette approximation est bénéfique car elle permet de réduire un certain nombre de couplages comme le couplage du moment angulaire total et le moment angulaire orbital appelé le couplage de Coriolis dans lequel l’opérateur l2 est remplacé par ces valeurs propres l(l+1) dans l’ équation suivante: L’obtention de la matrice les sections efficaces des niveaux et des sous niveaux rotationnels de transition de vers j’ se fait par la même démarche développée avec CC. La réduction du nombre d’équations de couplage entre et a permis de réduire le temps de calcul. L’approximation soudaine d’ordre infinie (Infinite Order Sudden IOS) Cette approximation a été introduite par Park en 1974 et consiste à négliger la rotation des molécules lourdes (les énergies des niveaux rotationnels) à cause de leur masse ayant des constantes rotationnelles faibles avec des énergies très grandes pour pouvoir maintenir le système fixe lors de l’interaction. Cela entraîne une approximation du nombre d’onde .
Cette équation ne dépend plus de j et en suivant la même démarche développée dans les méthodes précédentes pour l’obtention de la matrice de transition et les sections efficaces, les conditions aux limites de la fonction radiale Fl ( R , β) de cette équation nous permettent de définir la matrice de transition Sl (β) . Les sections efficaces de transition de j vers j’ sont obtenues par le biais de la matrice T l (β) qui est donnée par la relation suivante: Les taux de collisions sont définis comme étant la moyenne thermique des sections efficaces. Ils sont aussi appelés les constantes de vitesse et sont obtenus par le produit de la densité de l’atome perturbateur avec la distribution de Maxwell moyennée des sections efficaces pour une température donnée. Les coefficients de vitesse de couplage étroit à spin libre pour la transition de u vers v sont sous cette forme:
La résolution des équations couplées du second ordre nous a permis le calcul des sections efficaces par la méthode du couplage fermé et les taux de collision par la distribution de Maxwell. Le calcul des sections efficaces et les taux de collision se fait grâce à un calcul numérique qui demande l’utilisation d’un logiciel appelé Code de MOLSCATS qui est écrit en Fortran et pratiqué pour les systèmes de collision comme les systèmes bâtonatome, toupie asymétriquebâton, atomediatome, atomerotateur rigide, rotateur rigiderotateur rigide, atometoupie (…). Ainsi nous avons choisi pour le cas de notre travail le système CNCNHe qui est de type rotateur rigideatome. La structure électronique de la molécule CNCN est desymétrieDans cette partie nous allons détailler les paramètres utilisés dans le code de MOLSCATS pour l’obtention des sections efficaces. La résolution des équations couplées du second ordre se fait dans ce code grâce à l’implémentation de SEP. Pour introduire la surface d’énergie potentielle (SEP) dans ce Code, deux méthodes sont possibles: le VSTAR et VRTP.