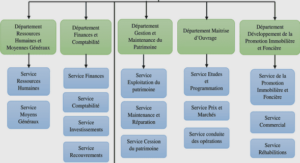
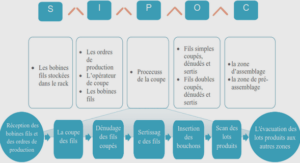
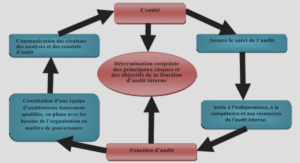
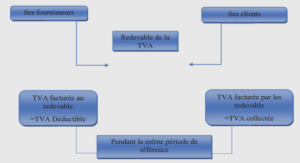
Echantillonnage
Cette étude concerne des adultes voiliers et non voiliers de C. maculatus qui ont déjà été échantillonnés au Sénégal, mis en élevage au laboratoire et conservés dans de l’éthanol après leur émergence. Tous provenant du Sénégal, nous avons défini comme population, tous les individus ayant la même capacité voilière. Tout compte fait, nous avons utilisé CmV pour coder les individus de la forme voilière et CmNV pour la forme non voilière. Première étape des manipulations au laboratoire, l’extraction de l’ADN des individus de C. maculatus a permis d’isoler l’ADN sous forme de solution aqueuse qui sera utiliser dans les étapes suivantes. Cette extraction a été réalisée en utilisant la trousse d’extraction QIAGEN DNeasy tissue et en suivant le protocole d’extraction standard des insectes (voir annexe 1). La tête, le thorax et les pattes de l’insecte sont seulement utilisés pour l’extraction de l’ADN. A la fin, pour vérifier la qualité totale de l’ADN extraite, une migration électrophorétique sur gel d’agarose 1,5% d’un mélange de 7 µl d’ADN et de 3 µl de bleu de charge est réalisée. Pour cela, le mélange est déposé dans les puits creusés avec des peignes lors du moulage dans le gel d’agarose. Ce dernier se trouvant dans la cuve remplie de solution tampon est exposé à une tension de 100 volts pendant 35 minutes. La révélation est faite après avoir laissé tremper le gel d’agarose dans du bromure d’éthidium pendant 30 minutes et rincer à l’eau par la suite avant la fluorescence sous lumière ultraviolet dans l’e-box V2®.
La réaction de polymérisation en chaine ou PCR permet de générer un nombre considérable de fragments identiques à la séquence cible facilitant ainsi le séquençage. Cette technique d’amplification in vitro de l’ADN repose sur la répétition de trois étapes : la dénaturation de l’ADN, l’hybridation des amorces sur la matrice et enfin l’élongation 3’ du brin néosynthétisé par la Taq polymérase (une enzyme thermophile). Cette réaction se fait en présence de l’ADN Pour notre travail, le gène d’intérêt est le cytochrome B, un gène mitochondrial. Gène à transmission maternel, le cytochrome B présente beaucoup d’intérêt dans les études visant à retracer l’histoire et la diversité des populations d’insecte. Il a été séquencé pour la première fois chez Sitophilus spp, ravageur des graines de maïs et c’est également révélé polymorphe et discriminant chez les insectes. Pour ce gène, les amorces utilisées sont le mtD26 (5′- TATGT ACTACCATGAGGACAAATATC-3′) et mtD28 (5′-ATTACACCTCCTAATTTATTAGGA AT-3′) (Barry et al., 2017) (Voir Annexe 2 et 3).
Le séquençage de l’ADN constitue une méthode dont le but est de déterminer la succession linéaire des bases A, C, G et T prenant part à la structure de l’ADN. Le séquençage se fait en suivant la technique de Sanger. Cette technique se fait en présence des composés habituellement utilisés lors de la synthèse du brin d’ADN lors d’une PCR en plus de didésoxynucléotides. Ces derniers sont des dNTPS dont le groupement OH a été remplacé par un groupement H et ont la particularité d’être couplés à des fluorochromes : ddATP-vert, ddTTP-rouge, ddCTP-bleu et ddGTP-jaune. Lorsqu’un ddNTP est incorporé à la place d’un dNTP, l’ADN polymérase ne peut plus continuer sa polymérisation. La réaction d’extension s’arrête car le ddNTP ne possède pas le groupe 3’-hydroxyle indispensable à la réaction de polymérisation. Statistiquement, au cours de la réaction, pour chaque « base » de l’ADN cible, au moins une fois, un ddNTP complémentaire sera incorporé à la place d’un dNTP. Par conséquent, à la fin de réaction, nous obtiendrons des fragments de tailles différentes. Lors de la migration, chaque fragment (contenant un ddNTP marqué par un fluorophore) sera excité par un laser et le signal obtenu sera analysé par un logiciel spécifique. L’analyse informatique des signaux permet d’obtenir la séquence étudiée, sous forme d’électrophorégramme de lecture manuelle aisée.
..multiple alignement (Thompson et al., 1994) du logiciel BioEdit version 7.2.5 (Hall, 1999). C’est une étape importante de l’analyse permettant de vérifier les éventuels gaps et mutations, de relever et corriger les erreurs éventuelles des séquences de chaque individus grâce aux chromatogrammes puis aligner l’ensemble des séquences. Cet alignement permet de ressortir les similitudes, les délétions, les insertions et/ou les substitutions. données global, de localiser les modifications pointues du cytochrome b de chaque forme et d’obtenir une estimation de leur polymorphisme. Ainsi grâce au logiciel DnaSP version 6.12.01 (Rozas et al., 2017), le nombre de séquences (n), le nombre total de sites (N), le nombre de sites invariables (i) et également le nombre de sites variables (v) ont été déterminés. Parmi ces derniers, sont distingués les singletons variables et les informatifs en parcimonie. Le nombre total de mutation (Eta) correspondant au nombre total de sites synonymes et non synonymes a été également déterminé avec DnaSP 6.12.01 (Rozas et al., 2017). Ce dernier a permis déterminer le nombre d’haplotype (h), le nombre moyen de différences nucléotidiques (k) de même que les diversités haplotypique (Hd) et nucléotidique (Pi) pour chaque population. Les taux de transversion (v), de transition (s) et de mutation (R), ainsi que les taux de mutations synonymes (Ks) et non synonymes (Kns) ont été calculés avec le logiciel MEGA version X (Kumar et al., 2018). Les changements nucléotidiques et d’acide animés sont ressortis avec le logiciel MEGA version X (Kumar et al., 2018).