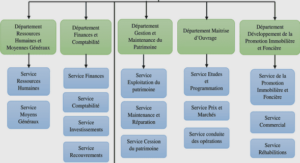
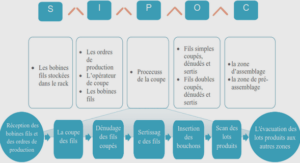
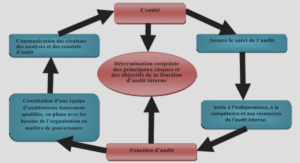
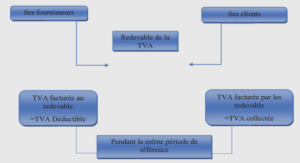
La métagénétique ou métagénomique ciblée pour étudier la diversité structurale
Une approche plus partielle pour décrire la structure des communautés est l’amplification et le séquençage d’un gène donné, généralement le gène codant pour la petite sous-unité 16S de l’ARN ribosomique (gène SSU rRNA), ARN non codant responsable de la traduction des ARN messagers en protéines (Klindworth et al., 2013). Ce gène présente l’avantage d’être ubiquitaire chez les procaryotes, de ne pas être sujet à des transferts de gènes horizontaux et d’être largement décrit dans les bases de données de référence (comme GenBank, SILVA ou encore EZ taxon). Il s’agit d’un gène d’environ 1500 pb (paires de base), présentant des régions conservées communes à tous les taxons et des régions variables (V1 à V9 ; voir Figure 4) résultant de mutations au cours de l’évolution et permettant de discriminer les différents taxons.
Le séquençage d’une seule région variable ne permet généralement pas de distinguer les taxons bactériens à l’échelle du genre ou de l’espèce, c’est pourquoi des fragments d’ADN contenant plusieurs régions variables sont généralement amplifiés à l’aide d’un couple d’amorces s’hybridant dans des régions conservées. Les séquences phylogénétiquement proches sont ensuite regroupées en Unités Taxonomiques Opérationnelles (OTU) assignées à des taxons biologiques. Pour le gène SSU rRNA, il a été défini que 97 % d’homologie de séquence constituait la limite de l’espèce, 90-95 % la limite du genre et 80-90 % la limite de la famille (Kim et al., 2014 ; Konstantinidis et Tiedje, 2005 ; Qin et al., 2014). L’assignation taxonomique s’effectue généralement par alignement de séquences sur le génome du représentant le plus proche disponible dans les bases de données. Dans le cas où les micro-organismes ne sont pas connus et/ou n’ont pas de représentant référencé, il peut arriver que l’affiliation ne soit possible qu’à des niveaux taxonomiques plus larges que l’espèce ou le genre (Werner et al., 2012).
Cette approche de métagénétique basée sur l’étude du gène SSU rRNA a largement bénéficié à l’étude des écosystèmes carnés (Benson et al., 2014 ; Chaillou et al., 2015 ; De Filippis et al., 2013 ; Fougy et al., 2016 ; Hultman et al., 2015 ; Jääskeläinen et al., 2016 ; Lauritsen et al., 2019 ; Li et al.,2019 ; Nieminen et al., 2012 ; Quijada et al., 2018 ; Raimondi et al., 2018, 2019 ; Rouger et al., 2018 ; Säde et al., 2017 ; Stellato et al., 2016 ; Zhao et al., 2015). Ces travaux seront présentés plus en détail dans la partie 1.2.2 Sources de contaminations possibles au cours des étapes de production. Dans le cas d’espèces phylogénétiquement proches, le gène SSU rRNA a un faible pouvoir résolutif lorsqu’il est utilisé sur des portions variables courtes et l’assignation est limitée au niveau du genre (Dahllöf et al., 2000 ; Head et al., 1998 ; Větrovský et Baldrian, 2013). Par ailleurs, le nombre de copies du gène SSU rRNA par génome varie d’une espèce à l’autre de 1 à 15 copies chez les bactéries (voir Figure 5) et jusqu’à 5 copies chez les archées, ce qui peut biaiser l’estimation des abondances relatives (Angly et al., 2014 ; Case et al., 2007 ; Crosby etCriddle, 2003 ; Větrovský etBaldrian, 2013).
Les indicateurs de diversité
Diversité intra-échantillon
La diversité intra-échantillon, également appelée alpha-diversité, mesure la diversité au sein d’un unique échantillon. Le nombre d’espèces présentes dans un échantillon donné constitue un indice de diversité alpha. Cet indicateur permettant d’estimer la richesse spécifique dans un échantillon a bénéficié à l’étude des produits carnés. Il a par exemple permis d’étudier l’évolution de la diversité au cours du stockage (Chaillou et al., 2015 ; Li et al., 2019 ; Raimondi et al., 2019), de comparer l’influence de différents traitements (Benson et al., 2014 ; Fougy et al., 2016) ou encore de comparer des échantillons issus de différentes zones de prélèvement (Stellato et al., 2016).
Diversité inter échantillon
La diversité inter-échantillons, également appelée beta-diversité permet de comparer les diversités respectives des différents échantillons deux à deux. Il existe deux grands types de distances : des distances compositionnelles et des distances phylogénétiques. La distance compositionnelle prend en compte uniquement les espèces présentes dans un échantillon, sans considérer leur lien généalogique, tandis que la distance phylogénétique prend également en compte l’éloignement phylogénétique des différentes espèces. Parmi les distances les plus utilisées pour comparer des communautés microbiennes, on peut citer la distance Bray-Curtis et la distance UniFrac. La distance Bray-Curtis (également appelée indice de dissimilarité de Bray-Curtis), mesure une distance compositionnelle entre les communautés bactériennes de deux échantillons. Cette distance tient compte de l’abondance des espèces. Elle correspond à la somme des écarts d’abondance de chaque espèce entre les communautés A et B, rapportée à la somme des abondances de ces espèces dans les deux communautés. Ainsi, plus les écarts d’abondance sont importants (communautés éloignées), plus la distance est grande.
Sources de contaminations possibles au cours des étapes de production
Sources de contamination de la viandecrue
Le muscle est stérile chez un animal en bonne santé (Elmossalami et Wassef, 1971), mais dès les premiers stades de la découpe, il entre en contact avec des micro-organismes. Il s’agit d’une contamination de surface par adsorption des bactéries (Chung et al., 1989 ; Luber, 2009). Différentes stratégies existent pour en identifier les sources : échantillonner directement sur le produit avant ou après une étape de production ou échantillonner l’environnement de production, incluant l’air, le liquide et les surfaces de travail.
Au cours des étapes d’abattage et de découpe des carcasses, des bactéries associées à l’animal peuvent contaminer la viande crue. Des bactéries telles que Propionibacterium acnes et Staphylococcus equorum, par ailleurs connues pour être associées à la peau des animaux ont par exemple été observées sur des steaks de bœuf, de veau, de volaille ou encore des dés de lardon en début de stockage (Chaillou et al., 2015 ; De Filippis et al., 2013). Dans le cas du porc, malgré une étape d’échaudage des carcasses après abattage, Wheatley (2014) a montré que la viande crue contenait encore 101 ufc/g des 106 ufc/g présentes initialement sur la peau des animaux. De plus, des bactéries connues pour être présentes dans les microbiotes intestinaux et environnementaux ont également été retrouvées dans des échantillons de produits animaux (Chaillou et al., 2015).
Etat de l’art
D’autres bactéries appartenant par exemple à la famille des Enterobacteriaceae, peuvent être présentes sur la viande. Comme précisé par le préfixe « entéro- », elles sont connues pour provenir majoritairement du tractus intestinal et sont un indicateur du niveau de contamination fécale. Elles sont en particulier apportées au cours de l’étape d’éviscération (De Filippis et al., 2013 ; Wheatley et al., 2014). Il a également été montré sur de la viande de porc et de bœuf que les parties antérieures (bajoues, poitrine et paleron) des carcasses suspendues par les pieds et recevant les liquides de découpe, présentaient une charge bactérienne plus importante que les parties postérieures (flanc et membres postérieurs) et étaient particulièrement riches en Enterobacteriaceae (De Filippis et al., 2013 ; Wheatley et al., 2014). De façon similaire, des bactéries du genre Prevotella et Corynebacterium connues pour être présentes dans le rumen, ont été recensées sur des steaks de bœuf ou de veau (Chaillou et al., 2015).
Nous comprenons ainsi que le microbiote du tube digestif ou encore de la peau de l’animal peut contaminer la viande. Or, comme le microbiote du tube digestif est susceptible de varier selon l’alimentation des animaux (Liu et al., 2012) et les bactéries de la peau peuvent varier selon leur habitat (Monsallier et al., 2012), des changements de pratiques d’élevage peuvent potentiellement avoir des répercussions sur le microbiote de la viande. Par exemple, sur un produit cru comme du carpaccio de bœuf à DLC issu d’une même usine de production, Lucquin (2012) a constaté une augmentation de la prévalence de Lactobacillus fuchuensis et L. carnosum par 2 et 1,5 respectivement au printemps ainsi qu’une augmentation de celle de Brochothrix thermosphacta par 6 en automne. Dans cette même étude, la prévalence de Lactobacillus sakei et Leuconostoc mesenteroides était presque 1,5 fois plus importante en été qu’en automne ou au printemps.
L’auteur a suggéré que ce changement était en lien avec des modifications des pratiques d’élevage à cette période de l’année (alimentation, environnement).
Etat de l’art
Janthinobacterium lividum, une bactérie d’altération majoritaire en fin de stockage, provenait des surfaces de la ligne de production de l’abattoir. Cette étude a souligné l’importance de l’environnement de production dans la contamination du produit fini car les lots de viande de volaille issus d’une même matière première et ayant suivi un même circuit de production de l’abattage à la fabrication du produit avaient des communautés bactériennes semblables en fin de stockage. De façon similaire, en comparant les communautés microbiennes de cuisses de poulet de différentes marques, Rouger (2018) a constaté que des cuisses de poulet de deux marques différentes mais provenant d’un même abattoir et ayant été produites le même jour présentaient des communautés bactériennes proches (avec des abondances relatives semblables de Klebsiella, Budvica et Pseudomonas). De plus, il a été montré que des bactéries comme Pseudomonas sp., B. thermosphacta, Psychrobacter sp., S. equorum présentes sur du bœuf à l’air libre après une semaine de stockage étaient également présentes sur les surfaces de production de l’abattoir (De Filippis et al., 2013). La présence de bactéries comme Bifidobacterium choerinum (par ailleurs connue pour être présente dans les fèces animales) dans des environnements de production de viande de porc, suggère une inter contamination de la viande et de l’environnement de production (Gavini et al., 2006). La Figure 8 illustre le fait que la plupart des genres bactériens (Streptococcus, Brochothrix, Pseudomonas, Acinetobacter ou Psychrobacter pour les plus abondants) détectés dans la viande de bœuf ou de porc le sont aussi dans l’environnement de production.
Evolution des communautés pendant le stockage
La charge bactérienneaugmente au cours du stockage
Pendant le stockage, l’augmentation du nombre de bactéries est associée à un phénomène de croissance bactérienne. Le terme de croissance bactérienne englobe à la fois l’augmentation du nombre de cellules (divisions cellulaires) et l’augmentation du volume cellulaire.
Une façon simple d’estimer le nombre de cellules vivantes cultivables, à savoir la charge bactérienne dans un échantillon, est le dénombrement. Il s’agit d’estimer le nombre de colonies bactériennes formées sur un milieu de croissance non sélectif à partir d’un inoculum de cet échantillon. Cette estimation suppose que chaque bactérie isolée est à l’origine de la formation d’une colonie, d’où le terme de ufc (Colony Forming Unit / Unité Formant Colonie). Cette estimation est valable si les cellules sont disjointes, et si l’inoculum est suffisamment dilué pour que les colonies bactériennes soient distinguables.
La diversité bactérienne chute au cours du stockage
En sortie de production, la viande (cuite ou crue) arbore une diversité de contaminants, correspondant principalement aux phyla des Firmicutes, Proteobacteria, Bacteroidetes et Actinobacteria (Chaillou et al., 2015 ; De Filippis et al., 2013 ; Raimondi et al., 2018 ; Zhao et al., 2015).
Toutes les études de métagénétique s’accordent pour dire que la diversité diminue au cours du stockage. Elle a été estimée au début du stockage sur différentes viandes (bœuf, porc, veau, volaille) à plus de 200 OTUs et chute en fin de stockage à DLC, ou à l’altération à environ 20 OTUs (Benson et al., 2014 ; Chaillou et al., 2015 ; De Filippis et al., 2013 ; Ercolini et al., 2011 ; Fougy et al., 2016 ; Hultman et al., 2015 ; Lauritsen et al., 2019 ; Li et al., 2019 ; Nieminen et al., 2012 ; Quijada et al., 2018 ; Raimondi et al., 2018 ; Säde et al., 2017).
En comparant les communautés microbiennes initiales de différents produits carnés et de la mer, Chaillou (2015) a observé qu’un noyau d’espèces était commun à tous les produits (voir Figure 11), qu’un noyau d’espèces était typique des produits carnés, et que d’autres espèces étaient spécifiques d’un type de produit (par exemple, spécifiques des dés de lardons). Au cours du stockage, la croissance de ces espèces (et particulièrement de celles issues du noyau d’espèces initial) est favorisée dans certains types de produits. Ainsi, les communautés bactériennes sont de plus en plus différentes selon les produits au cours du stockage. Par exemple, P. acnes a été retrouvé spécifiquement dans les filets de saumon altérés alors qu’il appartenait au noyau d’espèces initialement commun à tous les échantillons..