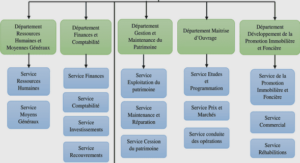
Exploration de l’expressivité nécessaire dans différents modèles biologiques
DR4 : Modélisation de sites de fixation de type DR4
Problématique
Nous faisons un retour sur l’application développée au chapitre 3 par l’intermédiaire des matrices. Nous montrons ici l’expérience de la modélisation grammaticale via le langage Logol. Dans cette étude, nous nous intéressons à la modélisation du site de fixation du facteur de transcription (Transcription Factor Binding Site, TBFS) de type « direct répété 4″ (Direct Repeat 4, DR4). Le DR4 est un motif répété avec un espace de 4 nucléotides entre les deux répétitions. Dans le cas de LXRα, qui est le facteur de transcription (FT) étudié par nos collaborateurs biologistes, le motif répété est un hexamère [MMYG06], ce qui donne la structure [Hexamère]NNNN[Hexamère]. Par ailleurs, dans les sites validés, la répétition n’est pas exacte et, en pratique, le second membre de la répétition diffère sensiblement du premier membre. L’objectif de la modélisation est de rechercher les séquences approchées de la séquence consensus d’un TFBS de type DR4, tel que celui de LXRα. Un autre objectif, plus ambitieux et à l’origine du projet, est de détecter les mots de type DR4 présents dans les séquences pour faire de la découverte de novo de sites de fixation. La capacité à modéliser une répétition constitue l’une des plus-values des grammaires SVG par rapport aux grammaires de plus bas niveau. Par ailleurs, Logol possède spécifiquement la capacité de pouvoir modéliser des répétitions approchées. Ce cas d’étude nous a donc semblé particulièrement adapté pour une modélisation par une approche grammaticale. Ces travaux ont fait partie des développements d’Orthocis présentés dans le chapitre 3. Ils ont été menés en collaboration avec Sandrine Lagarrigue et Frédéric Lecerf de l’équipe PEGASE de l’INRA à Rennes. Enjeux biologiques — Modéliser un TFBS de type DR4 (par exemple, LXRα) en vu d’une recherche pleingénome pour identifier des sites de fixation et prédire de nouveaux gènes ciblés. — Découvrir de nouveaux TFBS de type DR4, sans référence. Enjeu bioinformatique — Définir un modèle et un filtrage suffisamment spécifiques pour limiter les faux-positifs. Données disponibles Les références sont les 13 séquences de référence de TFBS de LXRα utilisées dans le chapitre 3 (cf. figure 3.1) et qui correspondent à des TFBS de LXRα avérés. Pour rappel, la séquence consensus associée à ces 13 séquences est : T GACCGnnnnT AACCC. Modèle Logol Le motif du TFBS LXRα a été modélisé sous forme de grammaire Logol (cf figure 5.1). Le modèle peut être subdivisé en 3 parties : le premier membre, le gap de taille 4 et le second membre. Chapitre 5. Exploration de l’expressivité nécessaire dans différents modèles biologiques 89 def :{ } mod1 () == > » TGACCG « :{ $ [0 ,3] , _PART1 }, GAP1 :{#[4 ,4]} ,? PART1 :{ $ [0 ,3]} mod1 () ==* > SEQ1 Figure 5.1 – Grammaire Logol pour le TFBS LXRα — Le premier membre est basé sur le consensus T GACCG, auquel on autorise jusqu’à 3 substitutions. En effet, parmi les références, le premier membre le plus éloigné du consensus compte deux différences (par exemple T GAACT sur le Cyp7Alpha1 chez la souris). Or, puisqu’on recherche les motifs approchés, il a été décidé de permettre une substitution supplémentaire. L’instance qui sera identifiée par la grammaire sera stockée dans la variable PART1. — Le gap est une séquence de taille 4 sans aucune contrainte de contenu. — Le second membre est l’instance sauvegardée dans PART1, pour laquelle on autorise jusqu’à 3 substitutions. En effet, dans les références, un second membre peut posséder jusqu’à 3 différences par rapport au premier membre (par exemple CGACCG/T AACCT sur le LPCAT3 chez l’humain). Ainsi, le modèle utilise le mécanisme de sauvegarde de l’instance : la troisième partie du modèle est comparée à la première partie telle qu’elle a été identifiée et non comparée au consensus. Cela permet d’exprimer la structure répétée « MnnnnM’ » avec M’ correspondant à M avec au plus 3 substitutions. Résultats biologiques Le modèle ainsi mis au point n’est pas spécifique de LXRα. En effet, le modèle est capable de reconnaître trop de séquences différentes, souvent très éloignées des séquences de références (par exemple : GGACGGacgtGGT GGG). La divergence observée au sein des références n’est finalement pas adaptée à une recherche de variants via des modèles grammaticaux (cf. chapitre 2). En conclusion, un modèle grammatical ne semble pas la meilleure stratégie pour rechercher des TFBS de type DR, surtout quand la répétition est assez déformée : chaque élément de la paire peut-être éloigné du consensus et les deux copies ne sont pas semblables. Réflexion sur le modèle Logol Pour rappel, l’intérêt initial de cette problématique était de modéliser des motifs de type DR4. Les DR étant des répétions directes d’un mot, une approche grammaticale avec une grammaire de type SVG semblait tout à fait indiquée pour les modéliser. Or l’approche a échoué sur le facteur LXRα. Le problème vient du fait qu’en pratique, le motif LXRα n’est pas un DR4 parfait, ce qui complique la modélisation. Ainsi, la répétition elle-même compte en moyenne deux à trois différences entre les deux éléments répétés. Par ailleurs, les références sont trop éloignées et ne permettent pas de définir un consensus solide. Ainsi, le premier membre ne compte que deux positions qui ne peuvent être mutées (le G en position 2 et le C en position 5).
Amorces mutées
Recherche d’amorces mutées dans des données métagénomiques
Problématique
Cette section détaille la modélisation des amorces mutées employées dans le chapitre 4. Elle s’inscrit dans le cadre d’une analyse métagénomique de la biodiversité dans des échantillons séquencés. Des erreurs peuvent survenir durant le séquençage, aboutissant à des reads avec des amorces mutées. Ces reads sont habituellement rejetés par le workflow d’analyse ([HHM+07]). La question biologique était de savoir si la détection d’amorces mutées pouvait permettre d’augmenter le nombre de reads analysables et si cela pouvait permettre d’identifier de nouvelles espèces présentes dans les échantillons. Notre objectif était de modéliser les amorces selon le modèle de mutation défini par les biologistes. Ce profil demandait un contrôle fin des paramètres du modèles, qu’ils ne réussissaient pas à obtenir avec les outils usuels. Nous avons donc essayé de modéliser leurs contraintes sous la forme d’un modèle grammatical via Logol. def :{ } mod1 () == >( » ccagca « :{ $ [0 ,2]} ,( » c « :{ $ [0 ,1]}| » g « :{ $ [0 ,1]}) , »c « :{ $ [0 ,1]} ,( » c « :{ $ [0 ,1]}| » t « :{ $ [0 ,1]}) , » gcggtaattcc « :{ $ [0 ,2]}) :{ $ [0 ,2]} mod2 () == >( » c » , » cagca « :{ $$ [0 ,1]} ,( » c « :{ $$ [0 ,1]}| » g « :{ $$ [0 ,1]}) , »c « :{ $$ [0 ,1]} ,( » c « :{ $$ [0 ,1]}| » t « :{ $$ [0 ,1]}) , » gcggtaattc « :{ $$ [0 ,1]} , » c « ) :{ $$ [0 ,1]} mod3 () == >( mod1 () | mod2 () ) mod3 () ==* > SEQ1 Figure 5.2 – Grammaire Logol pour l’amorce V4F mutée Ce travail a été effectué en collaboration avec Frédéric Mahé et Micah Dunthorn, du département d’écologie de l’université de Kaiserslautern, en Allemagne, et s’inscrit dans un projet de métabarcoding visant à faire l’inventaire de la biodiversité de sols de différents milieux tropicaux [MMB+15] (Costa Rica, Panama et Equateur). Données disponibles Les amorces exactes V4F & V4R Pour rappel (cf section 4.2.1), les amorces V4F et V4R sont respectivement décrites par les expressions régulières suivantes : CCAGCA [ CG ] C [ CT ] GCGGTAATTCC T [ CT ][ AG ] ATCAAGAACGAAAGT Amorce V4F mutée Les contraintes à modéliser étaient les suivantes : « 2 mutations ou 1 insertion/délétion possible. S’il y a une insertion/délétion, elle doit être interne (ie, ne pas affecter un nucléotide aux extrémités) » L’amorce V4F mutée a été modélisée avec une grammaire Logol (cf figure 5.2) où : — mod1 représente la grammaire V4F en autorisant jusqu’à deux substitutions ($[0,2]) — mod2 représente la grammaire V4F en autorisant jusqu’à une insertion/délétion ($$[0,1]) et les nucléotides aux extrémités ne sont pas sujet à la possibilité de mutations. — mod3 constitue l’axiome et permet la recherche de l’un ou l’autre modèle Dans le cas du modèle autorisant jusqu’à deux substitutions (« mod1″), chaque élément constitutif de la grammaire a reçu la possibilité de totaliser jusqu’à deux substitutions, à l’exception des positions alternatives de taille 1. Le contrôle total du nombre de substitutions est effectué par une vue (symbolisée par des parenthèses incluant l’ensemble des éléments constitutifs) qui a elle-même la possibilité de totaliser jusqu’à deux substitutions. Ainsi, la somme des substitutions des différents éléments constitutifs dans la vue ne doit pas dépasser 92 Chapitre 5. Exploration de l’expressivité nécessaire dans différents modèles biologiques def :{ } mod1 () == >( » t « :{ $ [0 ,1]} ,( » c « :{ $ [0 ,1]}| » t « :{ $ [0 ,1]}) ,( » a « :{ $ [0 ,1]}| » g « :{ $ [0 ,1]}) , » atcaagaacgaa « :{ $ [0 ,2]} ,( » agt « :{ $ [0 ,2]}| » ag « :{ $ [0 ,2]}| » a « :{ $ [0 ,1]}) ) :{ $ [0 ,2]} mod2 () == >( » t » ,( » c « :{ $$ [0 ,1]}| » t « :{ $$ [0 ,1]}) ,( » a « :{ $$ [0 ,1]}| » g « :{ $$ [0 ,1]}) , » atcaagaacgaa « :{ $$ [0 ,1]} ,(( » ag « :{ $$ [0 ,1]} , » t « ) |(( » a « :{ $$ [0 ,1]} , » g « ) ) | » a « ) ) :{ $$ [0 ,1]} mod3 () == >( mod1 () | mod2 () ) mod3 () ==* > SEQ1 Figure 5.3 – Grammaire Logol pour l’amorce V4R mutée le nombre de substitutions associées à la vue. Hormis cette restriction, toutes les répartitions des substitutions entres les différents éléments constitutifs sont possibles. Le même principe se retrouve pour contrôler le nombre d’indels dans le modèle autorisant jusqu’à un indel (« mod2 »). Les nucléotides à chaque extrémité du modèle n’ont pas reçu de possibilité d’indels, conformément aux spécificités des contraintes proposées par les biologistes. Amorce V4R mutée Les propriétés à modéliser étaient les suivantes : « 2 mutations ou 1 insertion/délétion possible. S’il y a une insertion/délétion, elle doit être interne (ie, ne pas affecter un nucléotide aux extrémités). Une absence d’un ou deux nucléotides terminaux est autorisée, sans que cela modifie les contraintes de mutation autorisées. » La dernière contrainte est nouvelle par rapport au modèle de mutation proposé pour V4F. L’amorce V4R mutée a été modélisée avec une grammaire Logol (cf. figure 5.3) où : — mod1 représente la grammaire V4R en autorisant jusqu’à deux mutations ($[0,2]) — mod2 représente la grammaire V4R en autorisant jusqu’à une insertion/délétion ($$[0,1]) et les nucléotides aux extrémités ne sont pas sujet à la possibilité de mutation. — mod3 permet la recherche de l’ensemble des modèles Le contrôle du nombre de mutations dans chaque modèle repose sur le même principe que pour la grammaire V4F (cf figure 5.2). La possibilité de tronquer la fin de la séquence a été modélisée par un choix représentant les différentes possibilités de fin : (« AGT »| »AG »| »A »), auxquelles ont été ensuite associées les possibilités de mutation. Conformément aux contraintes proposées par les biologistes, le dernier nucléotide n’autorise pas d’indels. Résultats biologiques Les résultats biologiques sont présentés plus en détail dans la section 4.3, page 79. Chapitre 5. Exploration de l’expressivité nécessaire dans différents modèles biologiques 93 Sur les échantillons 454/Roche, la recherche d’amorces mutées dans les données de séquençages permet d’augmenter le rappel de +8,3 % (25 619 amplicons supplémentaires détectés), pour un rappel total de 98,5 %. Sur les échantillons Illumina/MiSeq, la recherche d’amorces mutées dans les données de séquençages permet d’augmenter le rappel de +7,1 % (368 260 amplicons supplémentaires détectés), pour un rappel total de 89,8 %. L’analyse plus fine du devenir de ces séquences au terme du workflow d’analyse des biologistes a permis de démontrer que les séquences avec des amorces mutées ne sont pas elle-mêmes davantage mutées que les séquences avec des amorces exactes (similarité suffisamment conservée pour intégrer des clusters similaires). Par ailleurs, la détection des amorces mutées permet d’accroître la sensibilité des résultats en identifiant la présence de nouvelles espèces dans les échantillons séquencés (détection de +205 espèces potentielles en 454/Roche et +937 espèces potentielles en Illumina/Seq).