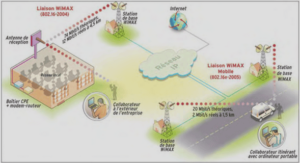
Elaboration et études des propriétés de films minces de carbone amorphe azoté (a-CNx) sur verre conducteur
La polyneuropathie amyloïde familiale à transthyrétine
La transthyrétine
La transthyrétine (TTR) est un biomarqueur dont les modifications post traductionnelles peuvent causer de graves maladies neurologiques comme la polyneuropathie amyloïde familiale . La TTR a été initialement appelée préalbumine (thyroxine binding préalbumine). En effet, lors d’une électrophorèse, le temps de migration de la TTR est plus petit que celui de l’albumine. Le site majeur de la synthèse de la TTR est le foie (95%), les autres sites étant le plexus choroïdes et la rétine. Cette protéine est donc présente dans le fluide cérébral spinal et dans le plasma (à une concentration de 0,20 g.L-1 à 0,40 mg.dL-1). Elle permet le transport de l’hormone thyroïdienne thyroxine et de la protéine de liaison du rétinol. Le chromosome 18 possède le gène qui code pour la TTR, ce gène est de petite taille et possède 4 exons et 3 introns. Le premier exon code pour un peptide et les 3 premiers acides aminés de la protéine. Le deuxième exon code pour les résidus de 4 à 47, l’exon 3 pour les acides aminés allant de 48 à 92 et de 93 à 127 pour le dernier exon [3]. La TTR normale est un tétramère soluble composé de 4 sous unités identiques. Un monomère est constitué de 127 acides aminés et chaque monomère à une masse de 14kDa. Les monomères sont structurés en feuillets béta plissés. Deux feuillets β composent un monomère et interagissent pour former un dimère, grâce à des liaisons hydrogène. Le tétramère contient deux sites de liaison de la thyroxine au centre de la molécule (Figure I-1).
Epidémiologie
La polyneuropathie amyloïde familiale (FAP) est une maladie à transmission autosomique dominante. Cependant environ 15% des personnes porteuses de la mutation sur le gène ne présenteront aucun symptôme. La FAP est caractérisée par des dépôts de substance amyloïde. Ces dépôts peuvent être dus à 3 protéines : la transthyretine TTR, la gelsoline et la lipoproteineA-1 [1][4]. La plus fréquente étant la polyneuropathie amyloïde familiale à transthyrétine. Cette maladie a été identifiée en 1952 au Portugal par Corino Andrade [5]. Cette maladie tout d’abord appelée amylose portugaise a par la suite été identifiée au Japon et en Suède. Ces trois pays sont de nos jours les principaux foyers de polyneuropathie à transthyrétine [6]. En 1978, Costa et al. découvrent que la transthyrétine est un constituant des dépôts amyloïdes. Il faut attendre 1983 pour que la première mutation de la TTR soit reportée. En 1989, 12 mutations sont connues, 60 en 1995, une centaine de nos jours (mutations simples ou doubles ou délétions dans le gène de la TTR). Parmi ces 100 mutations, 13 sont des mutations non pathologiques [7] (Figure I-2)Certaines de ces mutations sont non pathogènes comme la mutation Gly6Ser présente dans une population caucasienne à 12% [8]. Les mutations pathologiques provoquent la déstabilisation du tétramère entrainant la formation de fibres amyloïdes puis des dépôts Chapitre I Etude bibliographique 15 amyloïdes. Ces dépôts gênent le fonctionnement des organes touchés. Quelques études ont été réalisées sur le mécanisme d’amyloidogenèse de la TTR. Dans ces études, la première étape est la déstabilisation de la forme native de la TTR en raison des mutations amyloidogéniques. Ces mutations provoquent la formation de monomères. Ces monomères peuvent s’agréger et former des espèces oligomériques. Ces espèces peuvent par la suite former des fibrilles amyloïdes puis des dépôts amyloidiques. La formation de monomères est liée à la stabilité thermodynamique des formes de la TTR. La région entre des résidus 45 et 58 est le site de la plupart des mutations amyloidogéniques . La maladie touche des personnes de 18 à 83 ans avec un âge moyen de 35,3 ans. En 2012, en France, il a été recensé 386 patients atteints de la FAP. En France, celle-ci est causée par 29 mutations différentes, avec 66% des patients atteints de la FAP due à la mutation Val30Me.La mutation Thr49Ala située sur l’exon 3 est présente en France et en Italie. Cette mutation entraîne une perte de masse molaire de 30 g.mol-1. Cette mutation entraîne comme symptômes prédominants une neuropathie périphérique, une cardiomyopathie ainsi qu’un syndrome du canal carpien
Manifestations cliniques
Cette maladie se manifeste par des atteintes au niveau des nerfs, du cœur, du rein (10 à 20%) ou de l’œil. L’atteinte neurologique commence par se manifester au niveau des mains et des pieds, puis se développe dans tous les membres. Deux types de FAP existent : la FAP de type I (ou la neuropathie est seule), et la FAP de type II (il y a aussi un syndrome du canal carpien). D’autre part, il peut y avoir une atteinte du système nerveux autonome. Cette atteinte se manifeste par des problèmes gastro-intestinaux (diarrhées, constipation) et génitauxurinaire. Les personnes touchées par cette maladie peuvent aussi présenter des problèmes oculaires, tels qu’une opacité vitreuse, des glaucomes ainsi que des anomalies pupillaires. L’atteinte cardiaque, quant à elle, se manifeste par une insuffisance cardiaque et des troubles de la conduction cardiaque. Sur les différentes mutations du gène, quatre entraînent une atteinte oculaire ,onze une cardiomyopathie et dix une atteinte leptoméningée .
Traitement de la maladie
La prise en charge de la maladie s’effectue en traitant les symptômes et en limitant le développement de la maladie ainsi que par le traitement des symptômes. Le traitement pour stopper la maladie est la transplantation hépatique, la variante amyloidogénique de la TTR n’est alors plus fabriquée. La première transplantation dans le cadre d’une FAP a été pratiquée en 1990 en Suède. De nos jours plus de 1400 transplantations ont été effectuées. Pour 90% des patients transplantés, la FAP était liée à la variante VAm30Met. Après transplantation, une diminution des dépôts amyloïdes est observée. Dans le cas de patients greffés à un stade précoce de la maladie pour une mutation Val30Met, la progression de celle-ci est stoppée dans 70% des cas. Pour cette mutation, l’espérance de vie est multipliée par deux après transplantation. Cependant, la guérison n’est pas assurée. Cette évolution dépend de plusieurs facteurs, tels que l’âge du patient, la durée de la maladie, l’indice de masse corporelle modifié (produit de la masse corporelle sur la concentration en albumine), le stade de la maladie et le type de mutations. D’autre part, une greffe foie-cœur est parfois pratiquée sur un patient présentant une atteinte cardiaque. En effet, si seule la greffe du foie est accomplie le pronostic de réussite est mauvais, l’atteinte cardiaque évoluant après la greffe. De nos jours, 1% des patients ont eu cette double greffe. Pour des patients atteints de problèmes rénaux, une greffe rein-foie est faite (3% des patients). Pour les patients ne pouvant pas subir une greffe, des médicaments sont en cours de développement. Le but de ces médicaments comme le Tafamidis est d’empêcher la déstabilisation de la TTR en monomères et en intermédiaires toxiques (Figure I-3). D’autres axes de recherche biochimique sont également en cours de développement comme ceux de Pepys et al. qui ont développé des analogues aux substances P qui ralentissent le développement des substances amyloïdes.
CHAPITRE I. ETUDE BIBLIOGRAPHIQUE |