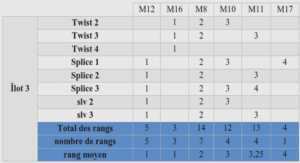
Description des SOFC
Le principe de fonctionnement des piles à combustible fut découvert en 1838 par Christian Friedrich Schoenbein, mais c’est Sir William Grove qui est considéré comme le père de ces systèmes. C’est lui qui réalisa en 1839 le premier prototype de pile et qui en fit une démonstration publique. Cette première pile utilisait l’acide sulfurique comme électrolyte et le platine comme matériau d’électrodes. Après ces premières recherches sur les piles, il n’y eut pas de développements importants pendant près d’un siècle. En 1953, Francis T. Bacon réalisa la première pile utilisant un électrolyte alcalin et des électrodes en nickel et oxyde de nickel. C’est ce prototype qui a convaincu la NASA d’équiper de piles tous les modules habités des programmes Gemini et Apollo dans les années 60. Ce sont encore aujourd’hui les piles à combustible qui sont utilisées dans les navettes américaines. Le choc pétrolier de 1973 a été un catalyseur pour le lancement de nouveaux programmes de recherches en Europe, au Japon et en Amérique du Nord. Cependant, dans les années 80, les recherches menées en Europe diminuent considérablement alors que le Japon et l’Amérique du Nord poursuivent leurs efforts. Il faut attendre les années 90, avec la prise de conscience des différents problèmes écologiques, la diminution des réserves en énergies fossiles et les progrès de la recherche, pour que de nouveaux programmes soient lancés en Europe.
Une pile à combustible permet de transformer l’enthalpie libre d’une réaction d’oxydation en énergie électrique. L’élément de base d’une pile à combustible est la cellule élémentaire constituée de trois couches :C’est au travers de l’empilement de ces trois éléments que la réaction d’oxydation a lieu. Le carburant, espèce réductrice, est approvisionné du côté de l’anode, alors que le comburant, l’oxydant, se trouve du côté de la cathode. Dans le cas des SOFC, c’est le comburant qui traverse l’électrolyte pour réagir avec le carburant. Pour ce faire, l’oxygène doit se réduire pour donner naissance aux ions oxonium qui peuvent traverser le conducteur purement ionique qu’est l’électrolyte :L’espèce électrochimiquement active, le comburant, n’est pas présente avec la même concentration du côté anodique et du côté cathodique. C’est cette différence d’activité chimique qui est à l’origine de la différence de potentiel électrique aux bornes d’une cellule.Il y a réduction de l’oxygène à la cathode (Réaction I.3) ; les ions ainsi créés diffusent à travers l’élec- trolyte afin de réagir avec le carburant à l’anode (Réaction I.4). Le principe de fonctionnement est schématisé par la Figure I.1.
En plaçant la cellule dans les conditions d’utilisation (température T, pressions partielles d’oxygène prespectivement à la cathode et à l’anode) mais en la laissant toutefois en circuit ouvert, le système cellule/ compartiments gazeux se trouve alors à l’équilibre thermodynamique. Il y a de chaque côté de l’électrolyte équilibre entre l’oxygène gazeux et les ions oxygène (Figure I.2).Pour qu’une réaction chimique ait lieu, il est nécessaire que les réactifs atteignent un certain niveau d’énergie : l’énergie requise est dite d’activation. Ceci se traduit, dans le cas des SOFC, par une polarisation d’activation ηqui reflète le phénomène de transfert électronique entre le conducteur ionique et le conducteur électronique. La polarisation d’activation peut être reliée à la densité de courant i, à la densité de courant d’échange iPour qu’une cellule soit pleinement efficace, les réactifs gazeux doivent alimenter en permanence les zones réactionnelles. Le type de diffusion gazeuse rencontré dans une électrode dépend de la taille de la porosité : lorsque celle-ci est élevée, les chocs molécule-molécule sont prépondérants et il s’agit alors d’une diffusion classique qui suit les lois de Darcy ; par contre, lorsque la porosité est de petite dimension, ce sont les chocs molécule-paroi qui deviennent majoritaires, et l’on parle alors de diffusion de Knudsen. Quand la vitesse de réaction devient trop importante, la diffusion des espèces gazeusesn’est pas assez rapide et la pression partielle dans les zones réactionnelles est alors plus faible que celle du mélange d’alimentation. Cette différence entraîne une polarisation de concentration, qui se traduit comme pour la polarisation d’activation, par une baisse de la tension aux bornes de la cellule. On peut relier la valeur de polarisation aux coefficients de diffusion des différentes espèces gazeuses, à l’épaisseur des électrodes et aux pressions partielles des gaz : on obtient alors deux expressions pour la polarisation de concentration, l’une pour l’anode.