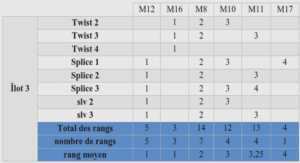
Arbres phylogénies
Ce chapitre présente les notions fondamentales permettant de comprendre les fondements et les difficultés de la reconstruction phylogénétique. Il est constitué de trois parties. La première partie introduit les définitions et les propriétés des arbres qui sont utilisés pour représenter des histoires évolutives. La seconde partie rappelle les propriétés et les définitions importantes concernant les données moléculaires. La troisième partie présente les modèles d’évolution les plus couramment utilisés pour modéliser l’évolution des séquences nucléotidiques. Depuis Darwin, les arbres sont utilisés comme support pour représenter l’aspect temporel de l’évolution et les regroupements d’espèces qui en découlent. Cette partie présente la terminologie relative à cette représentation, donne un aperçu de la combinatoire des phylogénies et précise les principales propriétés des phylogénies valuées. L’hypothèse fondamentale de la reconstruction phylogénétique est que l’histoire évolutive des espèces se déroule par spéciations successives. Suivant cette hypothèse, une lignée ancestrale peut, par spéciation, donner le jour à deux nouvelles lignées, et une phylogénie peut être représentée par un arbre similaire à ceux de la Figure 1. Ce paragraphe introduit plusieurs concepts qui sont fréquemment utilisés tout au long de cette thèse. Certaines des notions évoquées ci-dessous s’appuient sur des notions élémentaires de la théorie des graphes. Des définitions formelles de ces notions sont proposées par Berge (1970). 1 est un arbre dont les feuilles sont bijectivement associées aux séquences de E, et qui possède un seul sommet de degré 2 qui en est la racine.
Les feuilles de la phylogénie représentant les séquences contemporaines étudiées sont parfois appelées nœuds externes. Les autres nœuds de l’arbre, qui correspondent à d’hypothétiques séquences ancestrales des séquences étudiées, sont parfois appelés nœud internes. Un nœud interne peut aussi être vu comme un événement de spéciation. La racine de la phylogénie est le seul nœud interne de degré deux qui représente une spéciation. Les nœuds internes de degré supérieur à trois peuvent représenter le fait que plusieurs spéciations ont eu lieu et que l’on ne sait pas dans quel ordre elles se sont produites. Ils peuvent également représenter le fait qu’une seule spéciation a engendré plus de deux nouvelles espèces. On considère généralement que ce dernier phénomène est peu probable, d’où la définition suivante : une phylogénie enracinée est dite complètement résolue si tous ses nœuds internes, autres que sa racine, sont de Soit T une phylogénie contenant une branche (n1, n2) qui relie le nœud n1 au nœud n2. Si l’on retire cette branche de T, on obtient deux composantes connexes. Soit E1 et E2 les sous-ensembles d’espèces (les feuilles de T) appartenant respectivement à ces deux composantes : E1 | E2 constitue alors une bipartition de E.
On dit que la bipartition E1 E2 134 5 . L’ensemble des bipartitions induites par T caractérise complètement la phylogénie T (Buneman 1971). On peut donc comparer deux phylogénies en comparant leurs bipartitions. En particulier, toutes les phylogénies portant sur le même ensemble de séquences (ayant les mêmes feuilles) ont en commun les bipartitions induites par leurs arêtes externes ; ces bipartitions ne sont donc pas réellement informatives. Dans le cas où, comme dans la Figure 1.b, l’ensemble des espèces étudiées est E = { ,2, , , }Les méthodes de reconstruction phylogénétique reconstruisent généralement des phylogénies non-enracinées. Dans la suite de cette thèse, nous parlerons toujours, sauf mention contraire, de phylogénie non-enracinée. ayant ()i + 1 feuilles en greffant la feuille supplémentaire sur une branche quelconque de Ti. Les phylogénies ainsi obtenues sont toutes distinctes, et possèdent toutes deux branches de plus que la phylogénie Ti. Pour 2 séquences, il existe une seule phylogénie, et elle possède une seule branche.
Pour n ≥ 2 , le nombre de branches d’une phylogénie complètement résolue ayant n feuilles est donc 12 2Nous avons pour l’instant omis l’aspect temporel de la phylogénie. Cet aspect temporel peut être représenté par une valuation positive des branches de l’arbre, leurs longueurs, et on dira alors que la phylogénie est valuée. Dans une phylogénie valuée, la longueur d’une branche (n1, n2) représente la distance évolutive qui sépare n1 de n2 et la distance entre deux nœuds correspond à la somme des longueurs des branches qui sont sur le chemin reliant ces deux nœuds. Par exemple, dans la phylogénie de la Figure 1.b, la distance séparant les feuilles 1 et 3 est la somme des longueurs des trois branches (1, 6), (6, 7) et (7, 3) qui constituent le chemin qui relie ces deux feuilles. En particulier, pour chaque couple de feuilles i, j d’une phylogénie valuée positivement, on peut obtenir la distance dij qui sépare ces deux éléments de E. La mesure de distance d ainsi obtenue est appelée distance d’arbre.