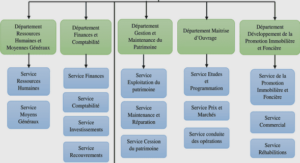
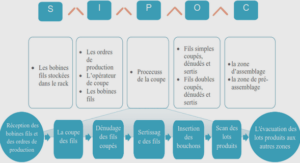
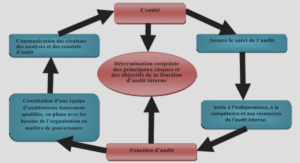
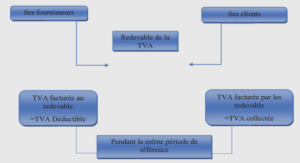
AGR fonctionnelle
La première étape a consisté à exprimer le besoin poursuivi par notre étude : la sécurisation du circuit du médicament expérimental. Comme le montre la figure ci-dessous, les bénéficiaires de ce service sont nombreux : patients, promoteurs, établissements de santé etc. Notre analyse devra tenir compte des risques liés à ces différents acteurs, bien que la priorité soit donnée à la sécurité du patient. (DM), bien que la sécurisation de leur circuit soit également un enjeu important, notamment pour les Dispositifs Médicaux Invasifs (DMI). Certaines observations faites dans notre étude peuvent s’appliquer aux DM, toutefois une seconde étude spécifique serait nécessaire L’objectif de notre étude est de sécuriser l’ensemble de la prise en charge médicamenteuse du patient, et non pas le seul circuit du médicament. Nous avons donc choisi d’élaborer un système sous forme d’actes, car cela permet de réunir les différents acteurs de la recherche clinique.
L’initiation de l’essai est une étape clé qui comprend notamment une pré-visite, une évaluation de la faisabilité et une visite de mise en place. La pré-visite ou visite de sélection permet au promoteur de vérifier l’éligibilité du centre investigateur. L’évaluation de la faisabilité de l’étude consiste à vérifier que chaque centre investigateur possède les moyens techniques et humains nécessaires au bon déroulement de la recherche, notamment lorsqu’une préparation magistrale est prévue. Cette évaluation de la faisabilité permet d’anticiper les contraintes organisationnelles éventuelles liées à la recherche. La visite de mise en place (MEP) a lieu au niveau du centre. Elle permet au promoteur de présenter les objectifs de l’étude et d’en expliquer son déroulement à l’équipe investigatrice. Le promoteur, en général représenté par l’ARC, s’assure que l’équipe investigatrice a reçu les informations nécessaires par le biais de documents officiels (protocole, brochure investigateur, notice d’information et formulaire de consentement…). L’investigateur principal se chargera de former et informer tout professionnel de santé participant à la recherche après avoir signé une délégation de tâches. Il peut déléguer à un TEC la rédaction des procédures opératoires standards (POS) et participe, conjointement au pharmacien, à l’élaboration de l’ordonnance type de l’essai.
Cette étape consiste à acheminer les traitements expérimentaux de la PUI au service de soins, lorsque le ME doit être administré dans le service. Le transport peut être effectué par le TEC de l’étude, l’IRC ou un coursier (cas des préparations de chimiothérapies). Dans les services d’Hospitalisation de Jour (HDJ), l’administration se fait dès réception du traitement. En revanche, dans les services d’hospitalisation classique, l’administration peut se faire plus tardivement : le traitement doit alors être stocké dans un endroit adapté. La clôture de l’essai a lieu en général lorsque le promoteur a un nombre suffisant de patients inclus dans l’étude pour satisfaire les exigences méthodologiques. L’essai sera clôturé lorsque tous les patients inclus seront parvenus à la fin du traitement. Une visite de clôture aura lieu dans chaque centre investigateur pour s’assurer que les données sont conservées conformément aux BPC et réévaluer les surcoûts a posteriori.
AGR système
Parmi les 26 dangers génériques proposés par l’ECP, le groupe de travail a sélectionné les DG impactant notre système, c’est à dire ceux pouvant conduire à des événements redoutés sur lesquels l’établissement pourrait mettre en place des actions d’amélioration. Les dangers génériques externes au système ne présentaient pas d’intérêt dans le cadre de notre étude, car les essais cliniques sont soumis aux mêmes dangers externes que la prise en charge classique. Notre cartographie regroupe 8 dangers génériques et 20 dangers spécifiques. Nous avons fait le choix de décomposer le DG « Facteur Humain » en fonction des différents corps de métier, au vus des fonctions très différentes de chaque acteur dans le processus.
Cartographie des situations dangereuses
Certaines situations dangereuses ont été regroupées lorsqu’elles concernaient plusieurs sous- fonctions de la même fonction (fusion horizontale). De même, d’autres situations dangereuses ont été regroupées lorsque, pour une même fonction et un même DG, plusieurs évènements dangereux étaient impliqués (fusion verticale). Ces modifications portent le nombre total de situations dangereuses à 137 : 56 SD de priorité 1, 30 de priorité 2 et 51 de priorité 10. L’AGR scénario a été élaborée à partir des 56 situations dangereuses jugées prioritaires. A partir de la cartographie des situations dangereuses et de l’analyse des risques a posteriori, une liste de scénarios d’accident a été élaborée et chaque scénario s’est vu attribuer une gravité et une vraisemblance, conformément aux échelles définies par le groupe de travail sur la base d’échelles de référence.
L’analyse des évènements indésirables déclarés en 2016 sur la base OSIRIS a révélé 294 événements indésirables en lien avec le processus de prise en charge médicamenteuse. Parmi ces évènements, 71 doublons ont été identifiés et 43 étaient inexploitables (description de l’évènement imprécise). Seulement 180 ont réellement pu être analysés, parmi lesquels 9 étaient sans incidence et 55 ne pouvaient pas être appliqués aux essais cliniques (ex : perte de la clé de l’armoire à stupéfiants entrainant un retard de prise en charge).