L’automédication antalgique chez les patients
drépanocytaires
L’hémoglobine
Structure
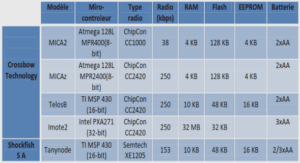
Structure primaire des chaînes d’hémoglobine L’hémoglobine est un tétramère de 67 000 Daltons. Elle est constituée de 2 chaînes α (composées de 141 acides aminés) et 2 chaînes β (146 acides aminés) de structures polypeptidiques (figure 1). Ces dimères sont associés de telle sorte que la sous-unité α1 est associée à la sous-unité β2 et l’α2 à la β1.
Structure spatiale
Structure secondaire Les différents segments des chaînes polypeptidiques adoptent dans l’espace des configurations stables. Les plus retrouvées sont l’hélice α et les feuillets β. L’hélice α est une spirale régulière constituée de 3,6 résidus par tour et possédant un pas de vis de 0,54mm. Cette hélice est stabilisée par de nombreuses liaisons hydrogènes (formées notamment entre les groupements CO et NH des liaisons peptidiques).
Structure tertiaire
Chaque chaîne de globine est repliée en 8 hélices (nommées de A à H, en partant de l’extrémité N-terminale) constituant une structure globulaire compacte. Cette structure est constituée à sa périphérie de résidus hydrophiles tapissant les chaînes latérales et en son centre de résidus hydrophobes constituant une poche qui accueille la molécule d’hème. L’histidine proximale est toujours en F8.
Structure quaternaire
Quatre sous unités s’associent pour former la molécule d’hémoglobine. A l’intérieur de ces quatre sous-unités, se trouve une poche qui sert à fixer l’un des métabolites de la glycolyse : le 2,3 diphosphoglycérate.
La molécule d’hèm
Une molécule d’hème est liée à chacune des sous unités. Elle est constituée d’une structure protoporphyrique contenant en son centre un atome de Fer (figure 1). La protoporphyrine est caractérisée par quatre cycles pyrroliques liés par l’intermédiaire de ponts méthylénés –CH= et substitués par des groupes méthyle, propionate et vinyle. 23 Le Fer est toujours à l’état réduit Fe2+. Lorsque la molécule d’hémoglobine est oxygénée, le Fer présente six liaisons de coordination : quatre sont liées à la structure de l’hème, une lie l’hème à la globine (au niveau de l’histidine F8) et la sixième fixe l’oxygène. Par contre, si la molécule d’hémoglobine est dépourvue d’oxygène, le Fer présente seulement cinq liaisons de coordination et le volume de l’atome augmente. Figure 1 L’hémoglobine Source : http://svtcolin.blogspot.fr
Fonction et effet coopératif
L’hémoglobine a pour rôle de transporter l’oxygène (O2). Dans le sang artériel, au niveau des poumons, la pression partielle en oxygène est élevée (de l’ordre de 100 millimètres de mercure (mmHg)) alors qu’au niveau du sang veineux, des tissus, elle est faible (de l’ordre de 40 mmHg). Pour de fortes pressions partielles en oxygène, l’hémoglobine va fixer la molécule d’O2 qu’elle libérera au niveau des tissus où la pression partielle en oxygène est plus faible. L’atome de Fer contenu dans le plan du noyau tétrapyrrole est légèrement surélevé en l’absence d’oxygène. Ainsi, lorsqu’il y a fixation d’une première molécule d’oxygène, l’atome de Fer va rentrer dans le plan défini par le noyau tétrapyrrole : ceci va déséquilibrer les hélices F et H des chaînes de globine ce qui va modifier les interactions entre les deux dimères α et β. Ces deux derniers vont glisser l’un sur l’autre ce qui va exposer les trois autres molécules d’hème à la surface de la molécule d’hémoglobine. Par conséquent, après fixation de la première molécule d’oxygène, les trois autres molécules d’oxygène vont se fixer beaucoup plus facilement : c’est l’effet coopératif. C’est ce qui explique la courbe sigmoïde de la saturation de l’hémoglobine en fonction de la pression partielle en oxygène (figure 2).
Génétique
La drépanocytose est une maladie causée par une mutation située sur le chromosome 11. Cette mutation est une substitution : une Adénine (GAG) est substituée par une Thymine (GTG) au niveau du 6ème codon du gène de la β globine. Ainsi un glutamate porté par l’hémoglobine A est substitué par une valine sur l’hémoglobine S. Cette mutation ponctuelle est à l’origine du polymorphisme d’un site de restriction nommé « restriction fragment length polymorphism » qui définit 5 haplotypes : 4 africains (Sénégal, Bénin, Bantou, Cameroun) et 1 arabo-indien. Cette mutation implique une nouvelle propriété de l’hémoglobine S : la polymérisation. En effet, le nouvel acide aminé hydrophobe (valine en position 6) remplace un acide aminé hydrophile. Ce nouveau site hydrophobe, au sein de molécules de globine entourées d’un film aqueux, devient alors un point d’ancrage entre deux molécules d’hémoglobine voisines (entre la leucine en position 88 ou la phénylalanine en position 85 d’une chaîne α et la valine 6 d’une chaîne β). Cette polymérisation survient en milieu concentré, ce qui est le cas du globule rouge. Par contre, quand la molécule d’hémoglobine S est oxygénée, il y a dissociation des polymères. Les patients présentant un syndrome drépanocytaire majeur sont homozygotes S/S ou hétérozygotes. Dans ce cas, ils sont porteurs d’une autre mutation de l’hémoglobine sur le deuxième allèle. Les autres mutations rencontrées sont : – L’hémoglobine C : c’est une mutation au niveau du 6ème codon du gène de la β globine où GAG devient AAG. Il y a ainsi substitution d’une glutamine par une lysine. La répartition de cette hémoglobine C est semblable à celle de l’hémoglobine S. Ces patients ayant un syndrome drépanocytaire majeur de type S/C ont une anémie moindre, une splénomégalie persistante au-delà de la petite enfance et des complications oculaires et osseuses plus fréquentes. – Bêta-thalassémies : il s’agit d’une mutation (souvent ponctuelle) au niveau du gène de la β globine. L’ARN messager codant pour la synthèse d’hémoglobine A au niveau des réticulocytes est soit présent en quantité insuffisante (thalassémie β+ ) soit non fonctionnel et toute synthèse de β globine est impossible (thalassémie β0 ). Les personnes atteintes des mutations S/β0 , ont une symptomatologie proche des patients ayant un syndrome drépanocytaire majeur de type S/S (homozygotes) alors que les personnes S/β+ ont une symptomatologie dépendante des taux d’hémoglobine A et/ou F (ne polymérisant pas avec l’hémoglobine S). Il existe des associations de l’hémoglobine S avec d’autres mutations de l’hémoglobine, un peu plus exceptionnelles telles que l’hémoglobine OArab ou l’hémoglobine DPunjab.
Physiopathologie
Polymérisation de l’hémoglobine
L’hémoglobine S, sous forme désoxygénée, se polymérise, déforme le globule rouge et lui donne ainsi une forme de faucille (figure 3). C’est sous cette forme que le globule rouge va perdre ses capacités de distorsion et d’élasticité dans le sang circulant (et notamment au niveau de la microcirculation) ce qui va entraîner un événement vaso-occlusif, caractéristique de la drépanocytose. Cette falciformation n’est pas immédiate : il existe un temps de latence (allant de la milliseconde à plusieurs minutes) qui correspond à l’agrégation des tétramères d’hémoglobine (Lionnet et coll., 2009). Une fois cet agrégat constitué et présent en quantité suffisante, il y a une croissance auto-catalytique, rapide, du polymère. Selon la concentration en désoxy-hémoglobine, le temps de latence est plus ou moins long. Ainsi, la présence de facteurs augmentant sa concentration ou stabilisant cette forme (acidose, hypoxie, fièvre, déshydratation) expose à des accidents de type vaso-occlusif. 26 Figure 3 Globule rouge drépanocytaire Source : Institut de Biologie Clinique – Hématologie CHU de Rouen Une autre composante influençant la falciformation est la concentration en hémoglobine. En effet, les α-thalassémies (parfois associées à la drépanocytose) diminuent les concentrations intra-érythrocytaires d’hémoglobine et réduisent la polymérisation. Le taux d’hémoglobine F est également à considérer car celle-ci ne polymérise pas avec l’hémoglobine S. La falciformation est réversible. Cependant la vaso-occlusion engendre un cercle vicieux (figure 4) en favorisant à son tour la polymérisation de l’hémoglobine (Wajcman et coll., 1992).
1 Introduction |