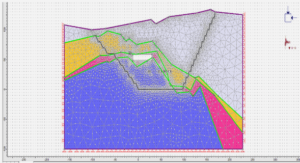
Extraction de réseaux de rues en milieu urbain à partir d’images satellites à très haute résolution spatiale
Polymères orientés
Outre la tendance naturelle qu’éprouvent certaines chaînes régulières à s’organiser en zones cristallines, il est possible de développer la cristallisation par une orientation mécanique des chaînes de polymères. Il résulte de cette orientation des polymères par étirage, une très nette amélioration des propriétés mécaniques, induisant une importance technique considérable. De plus, l’étirage de la matière provoque un accroissement très net du module d’Young selon l’axe de la fibre, une diminution tout aussi nette étant observée dans la direction perpendiculaire. L’élongation à rupture est réduite dans les deux directions. Sur le plan structural, on peut considérer que l’étirage provoque une réorganisation de la structure sphérolitique du polymère semi-cristallin et conduit à une orientation privilégiée des chaînes et des lamelles cristallines (Fig. 1.1). Fig. 1.1 Schématisation de l’effet de l’étirage sur la modification de structure d’un polymère semi-cristallin [FON94]. La température à laquelle est réalisé l’étirage joue un rôle prépondérant sur la qualité de l’orientation : l’étirage doit être impérativement effectué au-dessous de la température de fusion, afin d’empêcher la relaxation immédiate du système dès que la contrainte est relâchée.
Procédé de fabrication des fibres synthétiques
Les premières fibres synthétiques véritablement organiques (fibres de polyamide ou NylonTM, DuPont) sont apparues indépendamment aux Etats-Unis et en Allemagne vers la fin des années 30 [BUN96]. Puis les fibres de polyester se sont développées en Grande Bretagne dans les années 40, pour devenir les fibres synthétiques les plus produites. Depuis de nombreux développements ont permis d’accroître les propriétés mécaniques des fibres qui désormais rivalisent avec les meilleures propriétés mécaniques des aciers mais avec un gain de poids considérable (1/5). Pour accéder à de telles propriétés, deux approches ont été adoptées : d’abord, les structures moléculaires des fibres organiques ont été rendues plus complexes et par conséquent plus raides. La deuxième approche a été d’aligner la structure moléculaire des polymères simples pour améliorer considérablement les propriétés mécaniques. Beaucoup de fibres, particulièrement les fibres organiques, sont produites par étirage-filage suivi d’une opération de bobinage [BUN96]. Le processus d’étirage réduit le diamètre des filaments de 1 ou 2 mm à quelques microns. Il permet de modifier l’arrangement chaotique des molécules longues en une orientation ordonnée suivant l’axe des fibres. Ce changement de forme induit immédiatement des conséquences sur la résistance et le module d’Young des fibres.
Les fibres PA66.
En septembre 1931, le chimiste américain Wallace Carothers a fait part à la communauté scientifique de ses recherches dans les laboratoires de DuPont Company sur les molécules “géantes” appelées polymères [FIB03]. Le Nylon ou la “fibre miracle”, plus connu alors sous le nom de “66”, un numéro dérivé de sa structure moléculaire, était né. Le Nylon 6,6 est synthétisé à partir de la réaction entre l’hexaméthylène diamine et l’acide adipique qui permet d’obtenir l’adipamide diammonium hexaméthylène ou “sel de Nylon” [HU00]. Celui-ci est obtenu par réaction d’une solution à 70% méthanolique de diamine avec une solution à 20% méthanolique du Chapitre 1. Etude bibliographique 6 diacide. Le sel de Nylon précipite à mesure qu’il est produit. Pour extraire le poly(hexaméthylène adipamide) ou Nylon 6,6, la solution aqueuse à 60% de sel de Nylon est polymérisée. La polycondensation complète est représentée par la réaction suivante : HOOC–(CH2)4–COOH + H2N–(CH2)6–NH2 –HN–(CH2)6–NH–CO–(CH2)4–CO– + 2H2O Acide adipique Hexaméthylène diamine poly(hexaméthylène adipamide) (Nylon 6,6) Les fibres Nylon 6,6, comme la plupart des fibres thermoplastiques, sont produites principalement par le procédé de filage à l’état fondu ; le filage à partir d’une solution concentrée n’est pas très répandu dans l’industrie. Du fait de sa stabilité à l’état fondu et de sa basse viscosité, le Nylon 6,6 peut être filé à grandes vitesses. Plus récemment, un nouveau procédé de fabrication combinant à la fois filage et étirages a été développé : les cadences de production peuvent ainsi atteindre des vitesses d’étirage de l’ordre de 5000-7000 m/min, rendant le procédé de fabrication très économique. De plus, ce procédé s’avère moins polluant et plus simple à mettre en œuvre pour des matériaux dont le point de fusion est bien défini. Pour obtenir des fibres de Nylon, le polymère est tout d’abord introduit dans une extrudeuse sous forme de granulés de Nylon puis fondu par extrusion (Fig. 1.2). Le polymère fondu est alors pompé vers une filière, en passant par d’une série de filtres. L’opération de filage, c’est-à-dire la transformation d’une masse visqueuse de polymère en un filament continu, est un procédé complexe qui conditionne en grande partie les propriétés physiques et mécaniques du matériau à 120 capillaires pour former des monofilaments. Dès la sortie de la filière, les monofilaments se solidifient immédiatement au contact de l’air. L’application d’un jet d’air transversal permet de compléter leur solidification. Les fibres sont ensuite recouvertes par une fine couche huileuse. L’ensimage garantit la lubrification des fibres de manière à faciliter leur passage dans les différents outils du procédé de bobinage et à éliminer l’excès de charges électrostatiques. L’opération de bobinage confère une orientation préférentielle aux molécules du polymère, ce qui est déterminant pour les propriétés mécaniques des fibres. La résistance et le module des fibres sont augmentés et l’allongement à la rupture est diminué. Pour la production de cordes de pneumatiques, ces propriétés s’avèrent primordiales. Le bobinage permet d’étirer la fibre de plusieurs fois sa taille initiale, grâce à des vitesses de rotation différentes des bobinoirs. A température ambiante, une déformation de 100 à 400% peut être obtenue, dépendant de la vitesse de filage ; une augmentation de la température lors de cette opération induit des déformations supplémentaires. Pour améliorer leur stabilité à la chaleur, les fibres sont traitées à la vapeur. Pour certaines applications, par exemple dans l’industrie textile, les fibres sont délustrées en ajoutant du dioxyde de titane pendant la polycondensation ou avant l’extrusion.
Les fibres PET
En 1941, deux britanniques, J.R. Whinfield et J.T. Dickson, ont réussi à obtenir des fibres de polyester, à partir des travaux préalablement réalisés sur ce matériau par Carothers. Les premiers travaux de ce dernier portaient à la fois sur le Nylon et sur le polyester ; mais pensant que le Nylon était plus adapté à la fabrication de fibres synthétiques, il avait abandonné ses recherches sur les polyesters. La fabrication industrielle des premières fibres de polyester remonte à 1947 par la société ICI, sous le nom de Terylène. Cependant, la société DuPont, soucieuse de rester dans la course, a obtenu des inventeurs une licence d’exploitation. Les polyesters sont des polymères contenant des groupes ester caractéristiques, –O–CO–, dans leur chaîne principale. Ils sont communément préparés par la réaction d’un acide dibasique avec un alcool dihydrique [HU00]: HOOC–X–COOH + HO–Y–OH –OC–X–COO–Y–O– + H2O Acide dibasique Alcool dihydrique Polyester où X et Y sont des unités aromatiques ou alcalines. Dans le cas du poly(éthylène téréphthalate), PET, X est un anneau 1,4-phénylène et Y est éthylène. La polycondensation a lieu entre l’acide téréphtalique et l’éthylène glycol : HOOC– –COOH + HO–CH2CH2–OH –OC– –COO–CH2CH2–O– + H2O Acide dibasique Ethylène glycol poly(éthylène téréphthalate) (PET) Le procédé synthétique du PET peut être divisé dans deux étapes : • La première étape est, soit l’échange d’ester, soit l’estérification suivant le produit de départ. Si le diméthyle téréphtalate et l’éthylène glycol sont utilisés, la première étape est un échange d’ester, où le méthanol est distillé et un oligomère linéaire, bis(hydroxyéthyle)téréphtalate, est formé. Si l’acide téréphtalate et l’éthylène glycol sont utilisés, la première étape est un processus d’estérification, formant directement un oligomère linéaire, avec un dégagement d’eau. • La deuxième étape est un processus de polycondensation. Un catalyseur est ajouté au mélange des oligomères linéaires. Le glycol libre est distillé lorsque la polymérisation est effectuée à environ 285°C et à une pression inférieure à 25 Pa, jusqu’à l’obtention de la masse moléculaire désirée. Le catalyseur le plus utilisé pour cette étape est le trioxyde d’antimoine, mais le pentoxyde d’antimoine ou le dioxyde de germanium peuvent aussi être utilisés. En général, le procédé de fabrication des fibres PET consiste en un filage à l’état fondu suivi de plusieurs étirages à 90°C (Fig. 1.2). Il existe deux procédés de filage du polyester en fonction de la vitesse de bobinage: (i) un procédé à vitesse relativement basse (~1500 m/min) produit des fibres avec peu ou aucune orientation et (ii) un procédé à vitesse élevée (~3200 m/min) produit des fibres partiellement orientées. Ensuite, deux techniques de fabrication sont possibles : la première sépare les étapes de filage et d’étirage ; la seconde, par un couplage filage-étirage, permet d’atteindre des vitesses de bobinage avoisinant 6000 m/min. Le filage des fibres induit une texture à faible orientation cristalline. Les opérations successives d’étirages orientent favorablement les microstructures et confèrent des propriétés mécaniques utiles. Le procédé implique le passage des fibres autour d’une série de bobines avec des vitesses de plus en plus élevées pour atteindre un certain degré de déformation (taux d’étirage). Même si les fibres de polyester peuvent être étirées à la température ambiante, pour obtenir une fabrication homogène des fibres, il est nécessaire de les étirer à une température supérieure à la température de transition vitreuse. Usuellement, la température d’étirage se situe dans la gamme de 90 à 100°C. Celle-ci est déterminée précisément en utilisant soit un manchon chauffant entre les bobines, soit une bobine chauffée. Les taux d’étirage se trouvent habituellement entre 1.5:1 et 6:1. Pour la fabrication des fibres haute ténacité, on applique des taux d’étirage plus élevés et le procédé d’étirage s’effectue en deux étapes : au cours de la première étape, les fibres sont faiblement étirées puis lors de la seconde étape, on applique un taux d’étirage d’environ 6:1 à une température légèrement plus élevée. Si certes le procédé d’étirage confère un certain degré de cristallinité aux fibres, celui-ci s’avère insuffisant pour stabiliser la microstructure des fibres et ainsi empêcher un éventuel retrait thermique. Il est souvent nécessaire de cristalliser les fibres en les plaçant sur une plaque chaude ou une bobine de chauffage à 140-220°C pendant le procédé d’étirage.
Caractéristiques et propriétés des fibres
Afin de permettre la comparaison des matériaux étudiés au cours de cette étude avec d’autres types de fibres, quelques données physiques et mécaniques rapportées dans la littérature sont fournies dans ce paragraphe. La connaissance de ces propriétés est nécessaire pour définir le matériau approprié pour une application donnée. Par exemple, les propriétés recherchées dans un pneumatique sont une haute résistance, stabilité dimensionnelle, résistance à la fatigue, stabilité thermique et une bonne liaison avec la matrice.
Fibres PA66
Les fibres PA66 présentent une grande flexibilité car la molécule du polyamide 6,6 est elle-même flexible et peut s’orienter à chaque liaison [BUN96]; elles ont aussi une haute résistance à la traction, à l’abrasion, à la chaleur et possèdent une bonne stabilité dimensionnelle. La résistance à la traction des fibres PA66 est généralement affectée par les conditions et temps de chargement, le degré d’orientation moléculaire, la température et l’humidité relative [HU00]. Par exemple le taux d’humidité s’avère être un facteur plus pénalisant sur la propagation des fissures que sur l’évolution de la charge à rupture. Quant à l’effet de la température, il est plus significatif à un haut degré d’hygrométrie. La prise d’humidité du PA66 est typiquement conditionnée par la température ambiante et l’humidité relative. Celle-ci diminue avec l’incrément de l’orientation moléculaire et de la température. La recouvrance élastique des fibres est importante quand elles sont utilisées comme matériaux de renforcement, particulièrement pour des applications dans les pneus et pièces mécaniques en caoutchouc. Les fibres PA66 présentent une quasi totale recouvrance même à des charges élevées. Celle-ci dépend de la durée d’application de la charge et du temps permis pour la récupération. Quand le PA66 est exposé à la lumière solaire, il subit une dégradation qui provoque une perte de résistance. La photodégradation est oxydante, car la fibre est beaucoup plus stable dans l’azote. Un incrément de la température accroît la photodégradation, tandis que l’humidité n’a pas de grande influence. La présence d’un pigment augmente la dégradation, spécialement avec de l’oxyde de titane.
Fibres PET
La molécule de polyester contient des noyaux aromatiques la rendant moins flexible que la molécule de polyamide [BUN96]. La conséquence de cette rigidité moléculaire accrue est une augmentation du module des fibres de polyester, comparées aux fibres de polyamide. Le point de ramollissement des fibres PET est approximativement 260°C [HU00]. À environ 180°C, la ténacité des fibres PET est d’environ la moitié de sa valeur à la température ambiante. Les fibres PET ont une excellente résistance à l’oxydation et à la réduction. Elles résistent mieux au blanchissement sévère que d’autres fibres textiles. Les fibres PET ont une haute résistance à la plupart des solvants organiques et hydrocarbures communs. La gamme de produits chimiques qui dissolvent les fibres PET aux températures normales ou modérées est limitée. Les fibres PET ont une résistance à l’humidité considérablement élevée, dû à leur faible absorption d’eau (0.5 %). Les fibres PET présentent d’excellentes propriétés telles qu’une ténacité à rupture et un module élastique élevés, une résilience élastique modérée, des bonnes propriétés à la chaleur, une bonne résistance thermique et à la lumière [HU00]. Les tissus de fibre PET ont une bonne esthétique, une bonne résistance à la pliure, des bonnes propriétés de lavage et d’usage, et sèchent rapidement. Dû à leur excellente thermoplasticité (possibilité de thermofixage), les fibres PET retiennent le pli (elles ont été les premières fibres à être mises au point pour la réalisation de tissus à pli permanent). En plus, elles sont difficilement inflammables, sauf lorsqu’elles sont mélangées avec des fibres cellulosiques. Les produits de fibre PET régulier sont employés comme matériaux textiles. Les fibres PET industrielles sont classées en fibres de ténacité moyenne (MT), de haute ténacité (HT), de haut module (HM), de bas rétrécissement (LS) et les grades récemment développés de haut module/bas rétrécissement (HM/LS) ou les “dimensionnellement ” stables (DSP). Beaucoup des produits de fibres PET sont employés dans les matériaux composites renforcés avec des fibres. Parmi eux, les fibres HM/LS, récemment développées, sont largement utilisées dans les pneumatiques, les ceintures de sécurité, les bandes transporteuses et autres produits mécaniques utilisant une matrice de caoutchouc. Grâce à leur haute stabilité dimensionnelle, les fibres PET HM/LS ou DSP sont aujourd’hui les fibres les plus avancées pour le renfort de pneumatiques. Les fibres HM/LS offrent la même ténacité que les fibres Nylon de haute résistance. Leur retrait est typiquement de 6%, contre 8-12% pour le Nylon 6,6. Pour le renfort de pneumatiques, les fibres PET présentent une meilleure résistance à la fatigue et au “flat spotting”, par rapport aux fibres de Nylon. Ainsi, ce type de fibres PET s’avère plus approprié pour être incorporé sous forme de cordes dans les pneumatiques en caoutchouc, et ainsi fournir une meilleure résistance à la fatigue.
INTRODUCTION |