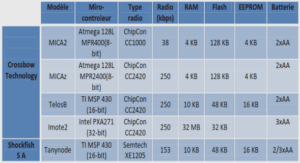
Aspects non usuels de la chimie des azaphosphatranes et proazaphosphatranes
La reconnaissance moléculaire
Présentation Les systèmes biologiques ont fourni une grande part d’inspiration au développement de la chimie supramoléculaire. De nombreux systèmes supramoléculaires synthétiques ont été conçus en effet pour mimer la structure ou le fonctionnement de processus biologiques complexes. Donald Cram avait déclaré qu’il était difficile pour des scientifiques familiers avec la chimie des systèmes biologiques d’éviter d’être inspiré par ceux-ci et que de nombreux chimistes organiciens admirant la structure cristalline des systèmes enzymatiques devaient rêver de penser et synthétiser des composés organiques plus simples qui imitent l’activité de ces enzymes.12 Le début de la chimie supramoléculaire, bien avant la conception de récepteurs sophistiqués tels que les carcérands, les cryptands et les systèmes qui s’auto-assemblent remonte au principe de la clé et de la serrure pour modéliser la catalyse enzymatique. Ce principe de sélection par la forme donne la première idée de ce qu’est un récepteur. Les premiers récepteurs furent synthétisés au début de l’ère de la chimie moderne. Le développement de la chimie supramoléculaire débuta par la synthèse de macrocycles au milieu des années 60, particulièrement par la synthèse de ligands macrocycliques pour les cations métalliques. Les éthers couronne ont été découverts en 1967 par un chimiste américain, Charles Pedersen13 travaillant chez Dupont de Nemours. La synthèse du premier composé de ce type se fit de manière accidentelle. Alors qu’il tentait de synthétiser le diol linéaire à partir du schéma réactionnel de la figure 1.6, le catéchol de départ dont l’une des fonctions hydroxy était protégée par une fonction THP était contaminé par une faible quantité de catéchol non protégé. Les produits de la réaction étaient alors un mélange du produit désiré et une faible quantité d’éther couronne. Figure 1.6 : Synthèse fortuite de l’éther couronne Dibenzo-18C6 Pedersen était alors intéressé par les propriétés de solubilité intéressantes de l’éther couronne synthétisé ainsi que le degré de cristallinité élevé qui suggérait qu’il s’agissait d’un composé moléculaire plutôt que d’un polymère. Le composé était faiblement soluble dans le méthanol mais sa solubilité était accrue de manière significative lors de l’addition de sels de métaux alcalins. trouva que ce composé était capable de dissoudre des sels inorganiques comme KMnO4 dans des solvants organiques comme le benzène et de donner une coloration violette à ce dernier. Il conclut finalement que le cation potassium était tombé dans un trou au centre de la molécule. Ce premier résultat très prometteur résulta en la synthèse rapide de toute une famille d’espèces à qui Pedersen donna le nom d’éthers couronne en raison de la forme de couronne du complexe moléculaire entre la molécule hôte et le cation K+ . Ces divers récepteurs peuvent complexer des cations métalliques de taille différente en fonction de la taille du macrocycle. Figure 1.7 : Exemple d’éthers couronne communs Peu après se sont développés les éthers lariat qui se démarquent des éthers couronne par l’ajout d’un bras flexible sur ceux-ci. Ils combinent ainsi la rigidité et la préorganisation des composés macrocycliques avec une stabilité additionnelle apportée par le bras introduit. Les cinétiques de complexation de cations métalliques sont également augmentées (figure 1.8). Le terme « lariat » fut donné par Gokel en référence à la forme de lasso qui est donnée à la structure finale. complexe deux cations K 15 Figure 1.8 : Complexation d’un cation métallique par un éther lariat Peu après le travail de Pedersen, Jean-Marie Lehn décida de concevoir des analogues tridimensionnels des éthers couronne. Des molécules bicycliques furent alors synthétisées, nommées cryptands en raison de leur capacité à entourer le cation comme dans une crypte (figure 1.9). Figure 1.9 : Exemple de cryptand En plus de Pedersen et Lehn, le prix Nobel de 1987 a été partagé avec Donald Cram pour son développement d’un autre type de macrocycles plus rigides, les sphérands (figure 1.10), pour la complexation de petits cations comme Li+ . Les groupements méthyle forment une surface lipophile sur la partie externe de la molécule tandis que les oxygènes des groupements méthoxy sont forcés de converger à l’intérieur de la molécule formant une poche pouvant accueillir un cation. Après ces premiers succès, de nouveaux récepteurs artificiels furent synthétisés permettant la complexation de molécules chargées ou neutres. On peut citer notamment les dérivés de cyclodextrines, les calixarènes, les cavitands et les cryptophanes (figure 1.9). Le développement important de ces nombreux récepteurs a démontré le désir des chimistes d’accéder à des récepteurs efficaces et sélectifs se rapprochant ainsi des enzymes. De plus, on peut accéder à des propriétés supplémentaires de ces récepteurs en les rendant hydrosolubles par l’ajout de bras hydrophiles ioniques ou de type PEG, par le développement de propriétés supramoléculaires macroscopiques par greffage de récepteurs sur supports solides ou polymères ou encore la création de sondes moléculaires par greffage de chromophores ou de sondes fluorescentes. Figure 1.10 : Structures illustrant les grandes familles de récepteurs Nous allons voir maintenant les caractéristiques que doivent posséder ces récepteurs pour en faire des hôtes efficaces.
Notions de récepteurs et complémentarité
Les récepteurs sont définis comme des structures organiques maintenues par des liaisons covalentes et capables de fixer sélectivement des substrats ioniques ou neutres au moyen d’interactions intermoléculaires diverses, aboutissant à un assemblage d’au moins deux molécules, une supermolécule. La reconnaissance moléculaire est définie comme l’énergie et l’information impliquées dans la fixation et la sélection d’un substrat par un récepteur moléculaire donné. La simple fixation n’est donc pas une reconnaissance. On peut ainsi dire que la reconnaissance est une fixation ayant un projet. Le processus de reconnaissance passe donc par un système hautement organisé par des interactions intermoléculaires. L’information est donc stockée à l’intérieur du récepteur. Celui-ci est caractérisé par sa taille et sa forme. Pour pouvoir interagir de manière O O O O O O H3C CH3 CH3 CH3 H3C CH3 sphérand O O O O O éther couronne O OH HO O OH OH O OH OH O O OH OH HO O O O OH OH HO O OH HO HO O O HO OH OH O cyclodextrine OH OH OH HO calix[4]arène O O O O O O O O R’ R’ R’ R’ R R R R cavitand O H3CO O O H3CO OCH3 O O O OCH3 OCH3 H3CO cryptophane A 17 efficace, il doit exister une complémentarité d’interaction entre les partenaires. Le concept d’adaptation stérique d’Emil Fischer recouvre des notions aussi bien géométriques qu’énergétiques. Il en résulte que l’enthalpie standard de réaction d’interaction entre un hôte et son substrat doit être fortement négative. On évalue l’interaction par une constante d’association Ka . Le principe de complémentarité avait été résumé par Donald Cram : « Pour complexer, les molécules hôtes doivent avoir des sites de coordination qui attirent de manière coopérative les sites de coordination des invités sans générer de fortes répulsions. » Voici quelques caractéristiques que doivent présenter les récepteurs : Complémentarité stérique : l’invité doit avoir une forme complémentaire à la cavité que présente le composé hôte. L’hôte doit s’adapter physiquement à l’invité sans être trop grand ni trop petit. La figure 1.11 montre les valeurs de constante d’association des éthers couronne avec différents cations. Li+ Na+ K + Rayon ionique (Ǻ) 0,60 0,95 1,33 12C4 <0,5 1,73 0,86 15C5 1,21 3,42 3,38 18C6 <0,5 4,32 6,15 Figure 1.11 : Structures de différents éthers couronne et valeurs de la constante d’association (données en log Ka) pour différents cations Complémentarité d’interaction : pour pouvoir se lier à un substrat, un récepteur doit posséder des sites de coordination qui soient complémentaires à ceux de la molécule invité. En d’autres termes, les caractéristiques électroniques (polarité, caractère donneur ou accepteur des liaisons hydrogène, dureté, mollesse…) doivent être complémentaires. A un site acide de Lewis du récepteur doit correspondre un site base de Lewis de la molécule invitée. En 1992, J. De Mendoza rapporta un récepteur possédant trois sites d’interaction complémentaires avec les sites d’interaction des acides aminés tryptophane et phénylalanine : un éther couronne qui peut interagir avec la partie ammonium de ces acides aminés, la partie guanidinium peut effectuer des liaisons hydrogène avec la partie O O O O O O O O O O O O O O O 12C4 15C5 18C6 18 carboxylate ainsi qu’une interaction électrostatique. Enfin des interactions de type π-π peuvent s’effectuer entre le groupement naphtalène et les noyaux aromatiques (figure 1.12). Figure 1.12 : Exemples de récepteurs combinant de multiples sites d’interaction Effet chélate et pré organisation : une des clés de la construction de récepteurs efficaces est la multiplication des sites d’interaction. Ceci signifie que l’on peut construire un complexe « hôte-invité » stable en utilisant des interactions non covalentes faibles si l’on s’assure qu’il existe autant d’interactions stabilisant le complexe que possible. Dans de nombreux cas, il existe une synergie entre toutes les interactions rendant le système plus stable globalement que la somme de toutes les interactions. Cette stabilisation supplémentaire trouve son origine dans l’effet chélate et macrocyclique. L’effet chélate est bien connu en chimie de coordination et relève de l’observation que les complexes métalliques comportant des ligands bidentate sont significativement plus stables que les complexes contenant des ligands unidentate3 . Si l’on prend l’exemple de la réaction ci-dessous, la valeur de la constante d’équilibre pour le remplacement de l’ammoniaque par le ligand 1,2-diaminoéthane indique que le complexe avec ce ligand est 108 fois plus stable. (Figure 1.13). O N O O O O O O N N N O O H H Cl N H CO2 – NH3 + CO2 – NH3 + Tryptophane Phénylalanine interactions interactions ioniques+ liaisons hydrogène interactions dipôle-ion complémentaires 19 Figure 1.13 : Equilibre en faveur de la formation du complexe chélaté La stabilité spécifique des complexes chélatés en solution a des origines à la fois cinétiques et thermodynamiques. Si l’on reprend l’équation de la figure 1.13, la réaction d’échange de ligand avec le ligand chélatant a pour conséquence une augmentation du nombre de molécules en solution et a donc une contribution entropique favorable. Deuxièmement, des effets cinétiques sont impliqués dans la formation du complexe chélaté. La chélation par un ligand L simple se fait à une vitesse similaire à la vitesse de coordination du premier site d’un ligand chélatant L-L. la coordination du second site se fait beaucoup plus rapidement parce qu’il est déjà lié au premier site et a donc une concentration effective beaucoup plus importante. La stabilité apportée par effet chélate dépend beaucoup de la taille du cycle formé. Les cycles à cinq chaînons sont souvent les plus stables pour des raisons de tension de cycle. De la même manière, la stabilité thermodynamique d’un complexe hôte-invité peut être augmentée par effet chélate. Cependant de nombreux systèmes supramoléculaires sont encore plus stables que la stabilité apportée par l’effet chélate seul. Les molécules hôtes impliquées comportent généralement des macrocycles qui chélatent les invités par des sites de coordination. De tels complexes sont stabilisés par l’effet macrocyclique. Les sites de coordination du récepteur n’ont pas, en effet, besoin de se rapprocher des sites de coordination de l’invité, ce qui empêche donc un coût enthalpique supplémentaire à « payer », c’est l’effet macrocyclique. 3 De plus, il y a moins de degrés de liberté qui sont perdus, empêchant une perte entropique supplémentaire. Par exemple, le sphérand de la figure 1.14 a déjà une structure pré-organisée alors qu’il y a un réarrangement conformationnel important lors de la complexation du cation K+ par le [2.2.2]cryptand (figure 1.14).
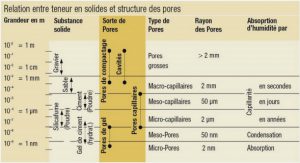
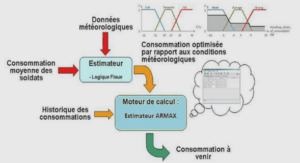
LISTE DES ABREVIATIONS |