Synthèses et études de complexes de ruthénium à ligands
Dans ce chapitre, de nouveaux complexes de ruthénium (II) similaires au black dye vont être étudiés. Les choix des routes synthétiques vont être définis selon les substituants souhaités en positions 4, 4’ et 4’’ sur le ligand terpyridine. Les études photophysiques et électrochimiques des nouveaux colorants permettront d’en établir les caractéristiques énergétiques pour définir leurs aptitudes à faire fonctionner les dispositifs photovoltaïques. La méthode de Hanan est idéale pour élaborer des terpyridines dites « symétriques », les fonctionnalisations seront au final identiques en position 4 et 4’’. Ces ligands seront ainsi à l’origine des complexes de ruthénium possédant deux fonctions d’ancrages homologues. Une première étude sera faite sur les groupes acides carboxyliques puis une seconde approche sur les acides cyanoacryliques. La première étape est la préparation des aldéhydes aromatiques. Ces intermédiaires ont été obtenus par O-alkylation avec un iodoalcane sur les substrats de départs préparés depuis la biomasse que nous avions privilégiés. 2 voies de synthèses ont été envisagées. La substitution nucléophile d’ordre 2 (SN2) est d’abord possible grâce à la déprotonation de la fonction alcool par l’hydrure de sodium dans le DMF à 0°C (Voie A, Figure 57). Des conditions différentes peuvent être utilisées pour la déprotonation des fonctions alcools par l’emploi de carbonate de potassium au reflux de la 2-butanone (Voie B, Figure 58). La 2-butanone est un solvant qui est plus facile à éliminer lors des traitements des différentes réactions.
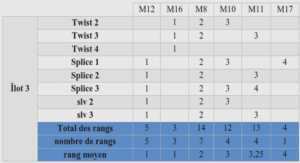
À partir de la vanilline, les rendements de la réaction de O-alkylation en passant par la Voie B approchent ceux de la Voie A (Tableau 1). En revanche, en établissant la réaction depuis le syringaldéhyde, de faibles rendements sont obtenus lors de l’utilisation du carbonate de potassium (5 % pour la méthylation et 13 % pour l’hexylation). Cela est dû à une acidité plus faible de la fonction alcool du syringaldéhyde par rapport à la vanilline. Une base forte comme l’hydrure de sodium est donc nécessaire afin de déprotoner le dérivé phénol. Les conditions réactionnelles utilisées à basse température (Voie A) sont donc privilégiées pour l’alkylation des dérivés de la biomasse. Les conditions ont été reproduites pour l’alkylation du 5-hydroxyméthylfurfural 3, un autre aldéhyde aromatique que nous avions choisi comme précurseur pour la formation d’une terpyridine. Cependant, le rendement établi pour l’introduction d’une chaîne hexyle est de 17 % seulement. Ce rendement a été amélioré à 50 % pour l’obtention du 5-((hexyloxy)méthyl)furan-2-carbaldéhyde 8 en descendant la température du bain à -15°C lors de l’ajout de NaH (Figure 59). La fonction ester en position 4 est à l’origine des futures fonctions d’ancrages CO2H des complexes de ruthénium (II) souhaités. Ce substrat de départ peut être accessible par acylation depuis le 4-isonicotinate d’éthyle. La méthode qui nous vient tout de suite à l’esprit est celle de Friedel et Craft, réaction catalysée le plus souvent par le trichlorure d’aluminium. Mais c’est sans compter sur le caractère π-déficitaire de la structure pyridinique qui empêche ce type de substitution électrophile aromatique de fonctionner (Figure 61).
Des conditions semblables à celles de Caronna et al. sont alors utilisées. L’introduction du groupe acétyle est catalysée par le fer (II) en présence de paraldéhyde, d’un acide et d’un peroxyde120 (Figure 62). Ces conditions opératoires permettent d’aboutir à une grande quantité du dérivé acétylpyridine mais le point faible reste la quantité importante de déchets générés par rapport à une acylation de Friedel et Craft (introduction de larges excès de paraldéhyde et d’hydroperoxyde de tert-butyle). Le radical tert-butoxyde est obtenu par réduction de l’hydroperoxyde de tert-butyle avec le fer (II). Cette étape entraîne par la même occasion la formation du fer (III). Il y alors abstraction d’hydrogène sur le paraldéhyde pour accéder aux radicaux acyles qui réagissent avec le noyau pyridinique protoné grâce à l’ajout initial de l’acide trifluoroacétique (TFA). L’aromaticité du cycle est régénérée par oxydation à partir du fer (III) sur les radicaux pyridiniques nouvellement formés (Figure 63). Ce cycle permet ainsi d’introduire une quantité catalytique du sulfate de fer (II) heptahydraté.