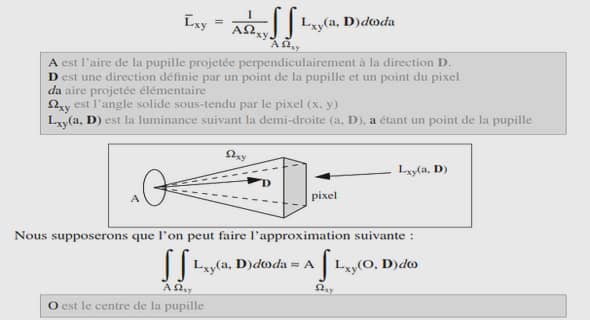
L’étude cinétique d’une réaction
L’étude cinétique d’une réaction nécessite de suivre l’évolution d’un signal représentatif de la concentration de l’un des réactifs ou de l’un des produits en fonction du temps. Les méthodes cinétiques sont donc qualifiées essentiellement en fonction de la résolution temporelle qu’elles permettent d’atteindre, c’est-à-dire l’ « âge » minimal observable d’une réaction. Parmi ces méthodes, on distingue principalement : – Les méthodologies en flux basées sur un mélange extrêmement rapide ; – Les techniques de relaxations basées sur la perturbation d’un système à l’équilibre par une variation brutale de température ou de pression ; – Les techniques de flash photolyse basées sur l’activation photo induite d’un proréactif ; – Les méthodes séparatives d’un état à l’équilibre vers un état hors-équilibre. Pour les techniques en flux, basées sur un mélange extrèmement rapide des réactifs, la résolution temporelle dépend notamment des facteurs cinétiques suivants : – Le temps d’injection des réactifs, τinj ; – Le temps de mélange des réactifs, τmix ; – Le temps mort du dispositif, c’est-à-dire le temps nécessaire aux réactifs pour atteindre le point d’observation τmin; – La durée d’acquisition du signal τaq; D’une part, τinj, τmix et τmin sont dépendants des techniques employées pour déclencher la réaction. D’autre part, τaq dépend de la méthode de transduction utilisée. Ces méthodes sont nombreuses, et dans le cas de l’étude des interactions entre un aptamère et une petite molécule, la cinétique du processus est le plus souvent suivie par spectroscopie de fluorescence ou par RMN. La somme de ces fenêtres de temps est limitante pour l’analyse cinétique d’une réaction. Pour être capable de mesurer avec précision une cinétique de réaction, il est impératif que la constante de temps de la méthode soit inférieure au temps de demi-réaction ⁄ , ou d’être capable de déconvoluer le temps de réponse global de l’appareillage du temps de réaction.
Technique en flux
Les techniques en flux sont, le plus souvent, couplées à une détection spectroscopique, et sont utilisées pour étudier des réactions sur des durées de quelques dizaines de microseconde à plusieurs centaines de secondes. De faibles volumes de réactifs sont rapidement poussés par des seringues vers une chambre de mélange, où la réaction s’initie. Puis ce mélange homogène gagne la chambre d’observation. La solution fraîchement mélangée n’est alors âgée que du temps qu’il lui faut pour passer de la chambre de mélange à la chambre d’observation, typiquement quelques millisecondes, c’est le temps-mort (τmin) du dispositif (Fig 60). Fig 60 : Schéma générale d’un mélangeur rapide couplé à de la spectroscopie. Les premiers dispositifs utilisant ces techniques en flux ont été décrits par B. Chance163 en 1964. Dans ce cas, le flux de mélange était alors ininterrompu (Continuous-Flow) et plus la mesure était effectuée proche du mélangeur, plus l’évènement observé était précoce. Il n’y avait dans ce cas pas d’exigences sur l’échantillonnage de la mesure, permettant l’utilisation de techniques d’analyses comme la spectrométrie Raman164,165 , ou la diffusion des rayons X aux petits angles166,167 . Les performances du Continuous flow ont été améliorées par M. Shastry168 en 1998 notamment en utilisant une caméra CCD (Coupled Charge Device, où le capteur est constitué de milliers de diodes photosensibles) et un mélangeur capillaire pour atteindre un temps de mélange de 15 µs et un temps mort de 45 µs. Ainsi, la solution est âgée de 60 µs lorsqu’elle arrive au détecteur. Il est alors impossible de mesurer un temps de réaction inférieur à cette limite. Les échantillons peuvent être analysés ici jusqu’à 1 ms après le mélange. Une alternative, pour consommer moins de matière, est le Stopped-flow, où lorsque le flux a gagné la chambre d’observation, il est stoppé à l’aide d’une troisième seringue (Stopping syringue), ce qui permet d’explorer une fenêtre de temps réactionnel allant jusqu’à la minute169 . La réaction est alors suivie dans le temps en enregistrant les variations d’un signal optique (généralement d’absorption ou de fluorescence) ou de conductivité. Les temps de mélange peuvent être réduits à la centaine de microseconde grâce à des mélangeurs turbulents de type Berger170, utilisés notamment par le constructeur OLIS et BioLogic. Ce dernier propose également des dispositifs où τinj + τmix + τmin< 0,6 ms. Ces dispositifs peuvent utiliser des volumes très faibles de l’ordre de la dizaine de microlitres dans la chambre de réaction, mais un minimum d’un millilitre est à prévoir pour remplir les seringues d’injection. Dans le contexte d’une reconnaissance entre un aptamère et sa cible, cette technique a été utilisée par J. Wachtveitl171 pour étudier la dynamique conformationnelle de l’aptamère anti-tétracycline, ainsi que le confinement de la tétracycline dans l’aptamère. Cette molécule est intrinsèquement fluorescente, et cette propriété est exaltée lorsque la tétracycline interagit avec l’aptamère. Un suivi Solution 1 Mélangeur Solution 2 Excitation Emission Absorbance Temps mort 63 de l’augmentation du signal de reconnaissance sera donc suffisant pour déterminer la cinétique de réaction. Disposant de la structure cristallographique du complexe Aptamère/Cible, ils ont également pu muter précisément quelques bases impliquées dans le site de reconnaissance ou dans le maintien de la structure tertiaire. Ainsi, ils ont montré que deux nucléotides étaient indispensables à une première étape de complexation, et qu’un troisième nucléotide, qui n’interagit pas avec la cible, verrouillait structuralement le complexe (fig 61). Cet exemple illustre les possibilités offertes par la méthode Stopped-flow d’accéder aux grandeurs cinétiques du processus de reconnaissance et d’identifier les étapes élémentaires du mécanisme.
Techniques de relaxation
Dans le cas des réactions trop rapides pour être étudiées avec ces méthodes de mélange, des méthodes dites de perturbation peuvent être utilisées. Dans ces techniques, il est question de perturber un système déjà à l’équilibre en appliquant une variation brutale d’un paramètre influençant l’équilibre, typiquement l’augmentation de la température (T-jump) ou de la pression 64 (Pressure jump). La cinétique de retour à l’équilibre ou l’établissement d’un nouvel équilibre est alors suivie. Dans le cas du T-jump, les sauts de température sont obtenus par effet Joule en faisant traverser un courant électrique à travers la solution. Classiquement, les sauts de température sont de l’ordre de 5°C en 1 à 2 µs. Les réactions de demi-vie plus courtes que la microseconde peuvent également être étudiées à l’aide de sauts de température induits par un laser en quelques nanosecondes 173,174 . L’inconvénient de cette méthode est que la température n’est pas maintenue en solution. Il existe donc une limite supérieure au temps d’étude de la réaction, de l’ordre de la dizaine de secondes. Cette technique permet notamment l’étude cinétique de repliement de protéines. En 1976, R. Clegg et B. Maxfield ont décrit le premier appareil à saut de pression. Cet instrument permettait d’appliquer des variations de pression de 0,01 à 5 atm. La subite variation de pression imposée à un système à l’équilibre altère l’énergie libre de réaction, dans laquelle il y a une différence de volume entre les réactifs et les produits : pour une réaction de type , si les réactifs ont un volume total V et le complexe un volume V + ΔV°, alors la réaction de complexation doit produire un travail PΔV° contre la pression extérieure pour se produire. Bien que les perturbations induites par des sauts de pression soient plus faibles que celles induites par un saut de température, la répétition de la perturbation n’est pas restreinte comme dans le cas du saut de température, où il faut attendre un retour à la température ambiante. Ce dispositif permet d’étudier des réactions de demi-vie > 20 µs, pendant plusieurs minutes. En 2002, D. Pearson177 rapportent un dispositif de Pressure-Jump en microvolume (50 µl) permettant d’appliquer des variations de pression de 200 atm, en 80 µs. Cette méthodologie a été appliquée à l’étude des interactions protéine-protéine ou protéine-ligand par spectroscopie de fluorescence. Le principe du Pressure-jump a également été adapté à de la diffusion des rayons X aux petits angles .
Technique de flash photolyse
La technique de flash-photolyse a été développée par R.G.W Norrish et G. Porter . au cours des années 1950, ce qui leur a valu le prix Nobel en 1967. Cette technologie était alors limitée aux réactions initiées par un pulse lumineux. L’applicabilité de cette méthode a ensuite été étendue à des méthodes dans lesquelles un réactif est emprisonné dans une cage moléculaire photosensible. A l’application d’un « flash », ces cages sont dégradées et libèrent le réactif dans un délai (temps de flash + temps de libération) de quelques ms à 200 ms. Ces techniques sont principalement utilisées pour l’étude de processus biologiques très rapides. Beaucoup de ligands photosensibles ont été mis au point pour interagir avec des nucléotides comme l’ATP , des neuro-excitateurs tels que le Lglutamate , ou encore pour chélater spécifiquement le calcium183 . Après photolyse, les réactifs sont relargués en solution et réagissent. Remarquons également qu’il est possible d’induire un changement brutal de pH par photolyse de o-nitrobenzaldéhyde184 . 65 Récemment, S. Pisch185 rapporte l’utilisation d’une guanine modifiée par un groupe nitrophenylethyl (NPE) photosensible qui permet d’empêcher un ARN de 20 nucléotides de coexister sous deux structures tertiaires différentes. La photolyse de la fonction NPE débloquant la guanine fonctionnalisée, l’ARN peut alors adopter la structure tertiaire inaccessible, et la mesure des paramètres cinétiques gouvernant la conversion d’une structure à l’autre peut alors être effectuée par RMN. Cette technique de flash photolyse est néanmoins limitée aux systèmes pour lesquels sont développés des cages moléculaires spécifiques ou aux molécules modifiables par une fonction photosensible qui bloque la reconaissance. Beaucoup de développements sont donc nécessaires pour chaque nouveau système.